Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(8):1571-1581. doi:10.7150/ijbs.34211 This issue Cite
Research Paper
Caffeine Inhibits NLRP3 Inflammasome Activation by Suppressing MAPK/NF-κB and A2aR Signaling in LPS-Induced THP-1 Macrophages
Weiming Zhao#, Li Ma#, Cheng Cai, Xiaohui Gong ![]()
Department of Neonatology, Shanghai Children's Hospital, Shanghai Jiao Tong University, Shanghai, China.
# These authors contributed equally to this work
Received 2019-2-18; Accepted 2019-5-2; Published 2019-6-2
Abstract

Excessive inflammation induced by various risk factors is associated with the development of bronchopulmonary dysplasia (BPD). Caffeine exerts potent anti-inflammatory effects as a clinical preventive medicine for BPD. Recently, NLRP3 inflammasome activation has been demonstrated to be essential for the pathogenesis of BPD. In the present study, we aimed to investigate the effects of caffeine on NLRP3 inflammasome activation in LPS-induced THP-1 macrophages and to explore the underlying the detailed mechanism. We found that caffeine significantly reduced NLRP3 expression, ASC speck formation, and caspase 1 cleavage and therefore decreased IL-1β and IL-18 secretion in THP-1 macrophages. Caffeine also markedly decreased the phosphorylation levels of MAPK and NF-κB pathway members, further suppressing the translocation of NF-κB in THP-1 macrophages. Moreover, silencing of the caffeine-antagonized adenosine A2a receptor (A2aR) significantly decreased cleaved caspase 1 expression in THP-1 macrophages by reducing ROS production. Given these findings, we conclude that caffeine inhibits NLRP3 inflammasome activation by suppressing MAPK/NF-κB signaling and A2aR-associated ROS production in LPS-induced THP-1 macrophages.
Keywords: Caffeine, NLRP3 inflammasome, MAPK, NF-κB, A2aR, THP-1 macrophages
Introduction
Bronchopulmonary dysplasia (BPD), the initial definition of chronic pulmonary insufficiency of prematurity [1], is a severe pulmonary complication with multifactorial pathogenesis [2]. Emerging evidence has indicated that multiple factors, including intrauterine chorioamnionitis, oxygen therapy, mechanical ventilation and nosocomial infection, lead to continuous injury in premature lungs that is mediated by inflammatory mechanisms [3-5]. Excessive secretion of proinflammatory cytokines including IL-1β predominantly by alveolar macrophages is thought to be responsible for the decreased alveolarization observed in BPD patients [6, 7]. Therefore, to be most effective, intervention to prevent or ameliorate BPD should focus on suppressing inflammation to protect the injured lungs [8].
In recent years, caffeine, which is extensively used for the treatment of apnea in preterm infants, has gained attention for its preventive effect on BPD [9]. Initially, researchers attributed the efficacy of caffeine in shortening ventilation time to its stimulation of the respiratory center, which further diminishes inflammation [10]. In addition, it has been reported that caffeine also exerts diuretic effects [11], provides neuronal injury protection [12] and induces surfactant protein B (SP-B) transcription [13]. Recently, growing evidence has suggested that caffeine can inhibit the secretion of proinflammatory cytokines, especially IL-1β, to reduce inflammation in hyperoxia and lipopolysaccharide (LPS)-induced rodent models [14-16]. However, the molecular mechanisms underlying the anti-inflammatory function of caffeine remain poorly understood.
A growing number of studies have revealed that activation of the nucleotide-binding domain and leucine-rich repeat protein 3 (NLRP3) inflammasome is related to the pathogenesis of acute lung injury [17, 18]. When macrophages sense external danger signals such as LPS, NLRP3 interacts with apoptosis associated speck like protein containing a CARD (ASC) and the precursor of caspase 1 to form the NLRP3 inflammasome [19, 20]. The activated NLRP3 inflammasome functions as a supramolecular platform for the caspase-1-dependent maturation and secretion of the proinflammatory cytokines IL-1β and IL-18 in macrophages [21, 22]. A recent study has shown that NLRP3-deficient mice exhibit reduced inflammation and improved lung alveolarization function under hyperoxia [23].
In addition, the hyperactivation of the NLRP3 inflammasome and elevated IL-1β levels in preterm infants with BPD also demonstrate the major role of NLRP3 in this condition [24]. However, it remains unclear whether caffeine inhibits NLRP3 inflammasome activation in macrophages under LPS stimulation. In this study, we aimed to assess the effects of caffeine on NLRP3 inflammasome activation and to explore the potential underlying mechanisms.
Materials and Methods
Cell culture and treatment
A human monocyte leukemia cell line (THP-1) was obtained from the Cell Bank of the Type Culture Collection of the Chinese Academy of Sciences. The cells were cultured at a density of 5×105 cells/mL in RPMI 1640 medium supplemented with 10% FBS and 1% penicillin/streptomycin solution (Biological Industries, Israel) at 37 ℃ in a 5% CO2 incubator. In all experiments, THP-1 monocytes were cultured in 6-well plates treated with 100 nM phorbol 12-myristate 13-acetate (PMA) for 24 h to transform into adherent macrophages.
Caffeine, ATP, LPS, PMA and Poly-L-lysine solution were purchased from Sigma-Aldrich (St. Louis, USA). Nigericin was purchased from InvivoGen (San Diego, USA).
Cytotoxicity assay
The cytotoxicity of caffeine toward THP-1 macrophages was evaluated by Annexin V-FITC and PI staining and flow cytometry (BioLegend, USA) according to the manufacturer's instructions. A total of 1×105 THP-1 macrophages were seeded in a 12-well plate and incubated with caffeine (100-800 μM) for 6 h.
ELISA for cytokine measurements
THP-1 macrophages were seeded in a 6-well plate at a density of 5×105 cells/mL. The cells were pretreated with caffeine (100-800 μM) or vehicle for 1 h and then treated with LPS (1 μg/ml) for 3 h and ATP (5 mM) for 30 min. The cell culture supernatants were centrifuged at 1500 rpm for 15 min at 4 ℃, and the pellets were discarded. Secreted IL-1β and IL-18 were measured by using commercially available ELISA kits (Abclonal, China) following the instructions provided by the manufacturers.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from cultured cells with TRIzol reagent (Life Technologies, USA) and reverse transcribed into cDNA using a Takara Prime Script RT Reagent Kit (Takara, Japan). qRT-PCR was performed using a TB Green PCR Kit (Takara, Japan) with a Roche Light Cycler 96 Real-Time PCR System (Roche, Switzerland). The PCR thermal cycling parameters were as follows: 95 °C for 600 s; 45 cycles of 95 °C for 10 s, 60 °C for 15 s and 72 °C for 15 s; and melting curve formation from 65 to 97 °C to ensure amplification of a single product. The GAPDH gene was used as an endogenous control. The relative gene expression was calculated using the 2-ΔΔCT method. The sequences of the primers used for qRT-PCR analysis are given in the table.
Primers used in this study
| Gene | GenBank accession no. | Primer sequence (5'-3') | Product size (bp) |
|---|---|---|---|
| GAPDH | NM_001115114.1 | F-GGACTCATGACCACAGTCCA R-TCAGCTCAGGGATGACCTTG | 157 |
| NLRP3 | NM_001079821.2 | F-AACATGCCCAAGGAGGAAGA R-GGCTGTTCACCAATCCATGA | 115 |
| Caspase 1 | NM_001257118.3 | F-GCACACGTCTTGCTCTCATT R-GCCTCCAGCTCTGTAGTCAT | 169 |
| ASC | NM_013258.5 | F-GTCACAAACGTTGAGTGGCT R-AAGTCCTTGCAGGTCCAGTT | 152 |
| IL-1β | NM_000576.2 | F-TGCTCAAGTGTCTGAAGCAG R-TGGTGGTCGGAGATTCGTAG | 189 |
| IL-18 | NM_001562.4 | F-TCGGGAAGAGGAAAGGAACC R-TTCTACTGGTTCAGCAGCCA | 96 |
| A2aR | NM_000675.6 | F-AGGCAGCAAGAACCTTTCAA R-CTAAGGAGCTCCACGTCTGG | 236 |
siRNA transfection
Gene knockdown experiments were performed in serum-free Opti-MEM using HiPerFect Transfection Reagent (Qiagen, Germany). Cells were transfected for 12 h with 5 nM human A2aR small interfering RNA (siRNA) (Qiagen, Germany) or irrelevant nontargeting negative control (NC) siRNA with Alexa Fluor 488 (Qiagen, Germany) or were treated with transfection reagent only (for mock transfection) according to the manufacturer's protocol. The cells were then further incubated for 18 h under standard growth conditions. At 36 h post transfection, depletion of A2aR was confirmed by qRT-PCR and western blotting.
Western blotting
Western blotting was performed using a standard protocol, as previously described [25, 26]. Briefly, total protein was extracted and separated by 10% SDS-PAGE using Mini-PROTEAN TGX gels (Bio-Rad, USA), and the separated proteins were transferred to PVDF membranes. The membranes were blocked with 5% nonfat milk at room temperature for 2 h and then incubated with specific primary antibodies overnight at 4°C. The primary antibodies, including antibodies against NLRP3, Caspase 1 (p20), P-p65 (Ser536), ERK, P-ERK (Thr202/Tyr204), JNK, P-JNK (Thr183/Tyr185), p38, P-p38 (Thr180/Tyr182), IκB-α, P-IκB-α (Ser32), cleaved Caspase 3 and ASC, were purchased from Cell Signaling Technology (Boston, USA). Antibodies against Caspase 1, Caspase 3, p65, A2aR, and GAPDH were purchased from Abclonal (Wuhan, China). Next, the membranes were incubated with appropriate secondary antibodies for 1 h at room temperature, and the bands were visualized with enhanced chemiluminescence (ECL) reagent (Thermo Scientific, USA). All the western blotting images for this study are representative of at least 3 independent biological experiments.
Immunofluorescence staining
For NF-κB staining, THP-1 macrophages were incubated with 800 μM caffeine or vehicle prior to treatment with LPS (1 μg/ml) for 3 h. Then, the cells were analyzed by immunocytochemistry as previously described [27]. The primary antibody against NF-κB p65 was from Abclonal (Wuhan, China).
The staining method used to explore ASC speck formation has been described in the literature [28]. Cells were incubated with 800 μM caffeine or vehicle prior to treatment with LPS (1 μg/ml) for 3 h and nigericin (10 μM) for 90 min. ASC specks were detected with a primary anti-ASC antibody (Abcam, UK). Images were captured using an Olympus FV1000 confocal microscope (Olympus, Japan) at 100× magnification. The nuclei were stained with DAPI (Beyotime, China). All the immunostaining images for this study are representative of at least 3 independent biological experiments.
Measurement of cellular reactive oxygen species
ROS production in THP-1 macrophages was detected using a Reactive Oxygen Species (ROS) Detection Assay Kit (BioVision, USA) and flow cytometry according to the manufacturer's instructions. The data were analyzed with CytExpert software.
Statistical analysis
The results are presented as the mean ± SD for at least three independent experiments. Student's t-tests were used for comparisons between two groups. Differences among more than two groups were analyzed using one-way ANOVA. Statistical analysis was performed using Graph Pad Prism 7.0 software, p< 0.05 was considered to indicate statistical significance.
Results
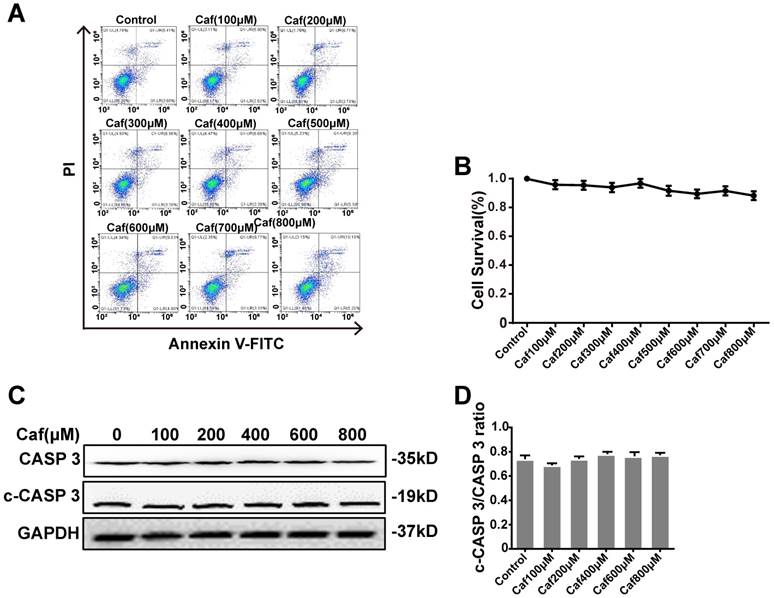
Effects of caffeine on cell survival and apoptosis
To test the cytotoxicity of caffeine toward THP-1 macrophages, we detected cell survival by flow cytometry. The results showed that none of the concentrations of caffeine had significant impacts on cell survival compared with the control (Figure 1A and B). To assess the effect of caffeine on apoptosis, we detected the cleaved Caspase 3/Caspase 3 ratio by Western blot analysis. The data indicated that treatment of THP-1 macrophages with caffeine did not alter the cleaved Caspase 3/Caspase 3 ratio at the protein level compared with the control treatment (Figure 1C and D). Afterward, we used 100, 200, 400, and 800 μM caffeine in the following experiments.
Effects of caffeine on THP-1 macrophage survival and apoptosis. (A) Cell survival was measured in THP-1 macrophages treated with caffeine (100-800 μM) for 6 h using Annexin V-PI staining and flow cytometry. (B) Bar graph showing the survival rates of all cell lines. (C) THP-1 macrophages were treated with caffeine (100-800 μM) for 6 h. The protein levels of cleaved Caspase 3 and Caspase 3 were analyzed by western blotting. (D) Grayscale analysis was used to quantify the relative expression of cleaved Caspase 3 and Caspase 3. The results represent the mean ± SD (n=3).
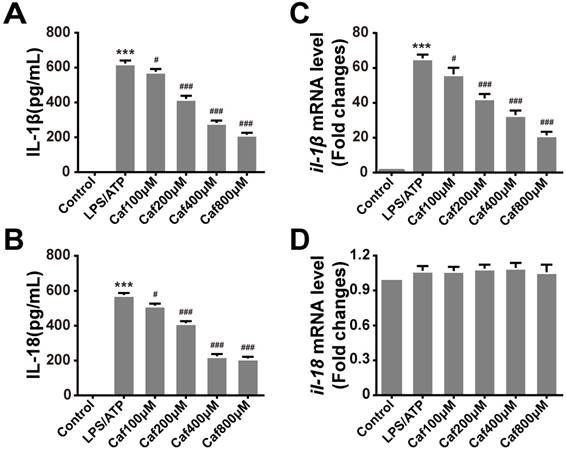
Caffeine suppresses IL-1β and IL-18 secretion in LPS/ATP-induced THP-1 macrophages
To test whether caffeine affects inflammasome-specific cytokine expression, we measured the secretion levels of IL-1β and IL-18 in the cell culture supernatants of LPS/ATP-induced THP-1 macrophages. The data showed that induction of THP-1 macrophages with LPS and ATP significantly increased the secretion of IL-1β and IL-18 (Figure 2A and B). Pretreatment with caffeine suppressed the release of both proinflammatory cytokines in a dose-dependent manner (Figure 2A and B). To further determine whether caffeine inhibits inflammatory cytokine expression, we detected the mRNA levels of IL-1β and IL-18. The results showed that LPS and ATP treatment increased the mRNA levels of IL-1β and that this effect could be attenuated by caffeine pretreatment (Figure 2C). On the other hand, the mRNA levels of IL-18 were relatively stable; they were affected by neither LPS/ATP stimulation nor caffeine pretreatment, indicating the continuous expression of this cytokine (Figure 2D). Based on the above findings, we assumed that caffeine inhibited NLRP3 inflammasome-specific cytokine secretion and that this effect was achieved by suppression of IL-1β gene expression.
Caffeine attenuates production of the cytokines IL-1β and IL-18 in THP-1 macrophages stimulated by LPS and ATP. (A) IL-1β levels were evaluated in THP-1 macrophages pretreated with caffeine for 1 h and then incubated with LPS (1 μg/ml) for 3 h and ATP (5 mM) for 30 min. (B) IL-18 levels were evaluated in THP-1 macrophages pretreated with caffeine for 1 h and then incubated with LPS (1 μg/ml) for 3 h and ATP (5 mM) for 30 min. (C) The mRNA levels of il-1β were analyzed by qRT-PCR. (D) The mRNA levels of il-18 were analyzed by qRT-PCR. The results represent the mean ± SD for three experiments. *** p< 0.001 vs. the control group. # p<0.05, ## p<0.01 and ### p< 0.001 vs. the LPS and ATP group.
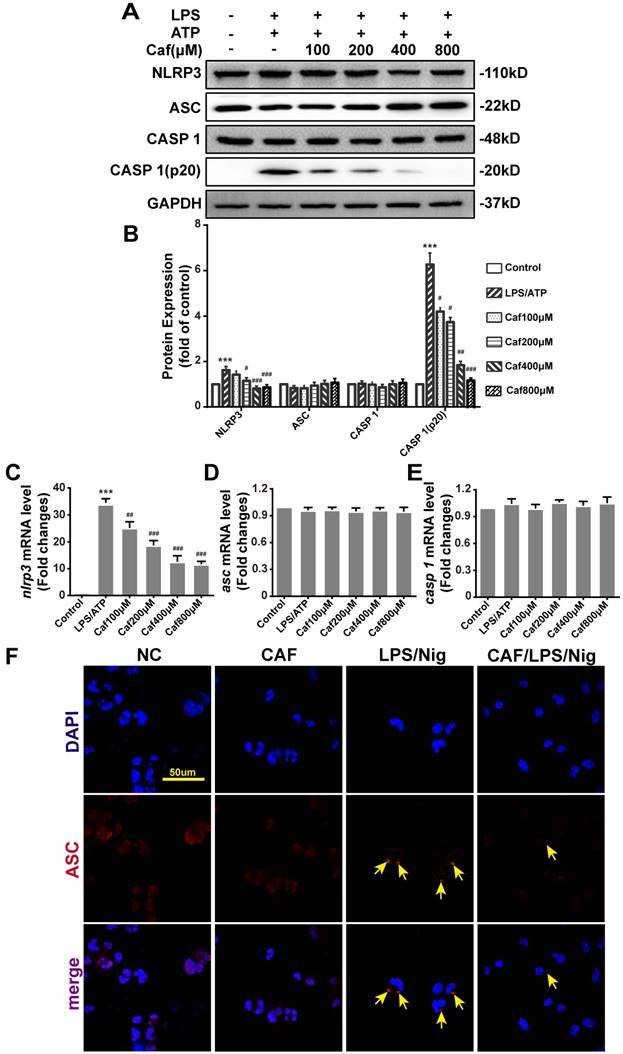
Caffeine attenuates the activation of the NLRP3 inflammasome
To verify whether the increased secretion of IL-1β and IL-18 under LPS stimulation contributed to activation of the NLRP3 inflammasome in THP-1 macrophages, we further examined the effect of caffeine on NLRP3 inflammasome activation. The results showed that upon LPS and ATP stimulation, the expression of NLRP3 mRNA and protein, which is the rate-limiting step for inflammasome activation [29], was significantly increased (Figure 3A-C). Moreover, the increased mRNA and protein expression of NLRP3 could be reversed by caffeine pretreatment (Figure 3A-C). The upregulation and activation of NLRP3 promoted Caspase 1 activation (Caspase 1-p20 levels), which in turn promoted the processing and secretion of IL-1β and IL-18. The PCR results also indicated that caffeine significantly reduced caspase 1 activation rather than Caspase 1 mRNA and protein expression (Figure 3A, B and E). When the NLRP3 inflammasome is activated, the adaptor protein ASC assembles into a large protein complex named an ASC speck or pyroptosome, which is considered a typical sign of inflammasome activation [30]. Our results showed that caffeine did not change ASC mRNA and protein expression (Figure 3A, B and D) but reduced the number of ASC specks stimulated by LPS and nigericin (Figure 3F). These observations suggest that caffeine can inhibit the activation of the NLRP3 inflammasome and the recruitment of ASC.
Caffeine inhibits NLRP3 inflammasome activation in THP-1 macrophages stimulated by LPS and ATP/nigericin. (A) The expression of NLRP3 inflammasome component proteins was evaluated by western blotting in THP-1 macrophages pretreated with caffeine for 1 h and then incubated with LPS (1 μg/ml) for 3 h and ATP (5 mM) for 30 min. (B) Densitometric analysis was used to quantify the levels of NLRP3 inflammasome protein expression. The mRNA levels of nlrp3 (C), asc (D) and caspase 1 (E) were analyzed by qRT-PCR. The results represent the mean ± SD for three experiments. *** p< 0.001 vs. the control group. # p<0.05, ## p<0.01 and ### p< 0.001 vs. the LPS and ATP group. (F) ASC speck formation was visualized by immunofluorescence analysis with an anti-ASC (red) antibody in THP-1 macrophages pretreated with caffeine (800 μM) for 1 h and then treated with LPS (1 μg/ml) and nigericin (10 μM) for 90 min. DAPI (blue) was used as a nuclear marker. Scale bar: 50 μm.
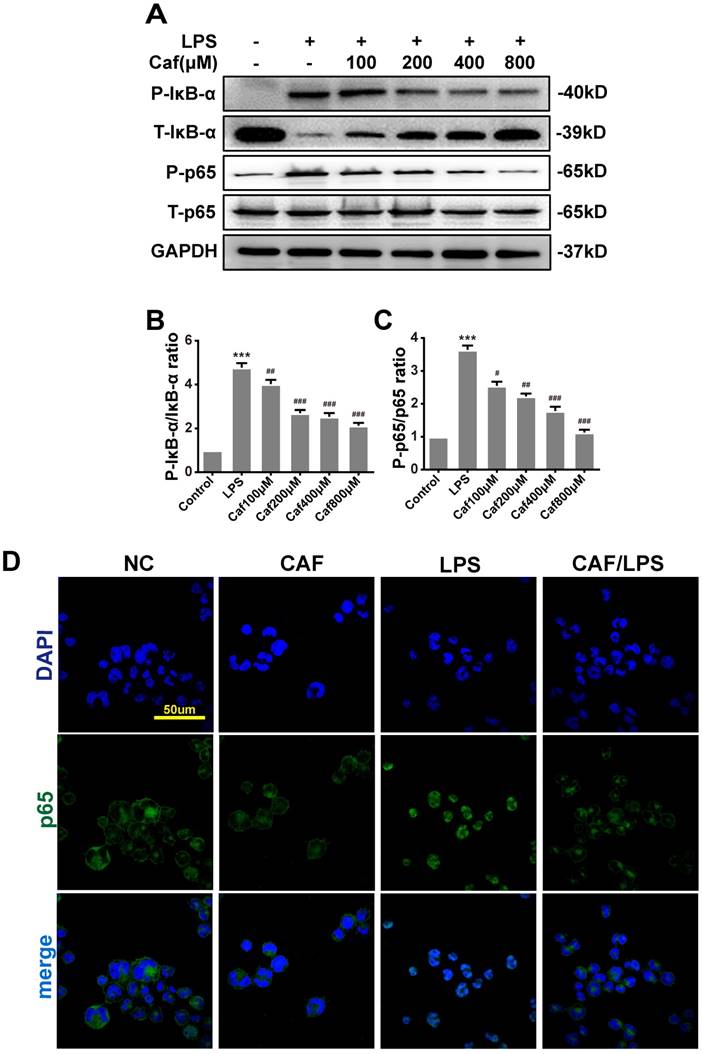
Caffeine inhibits NLRP3 inflammasome activity via the MAPK/NF-κB pathway
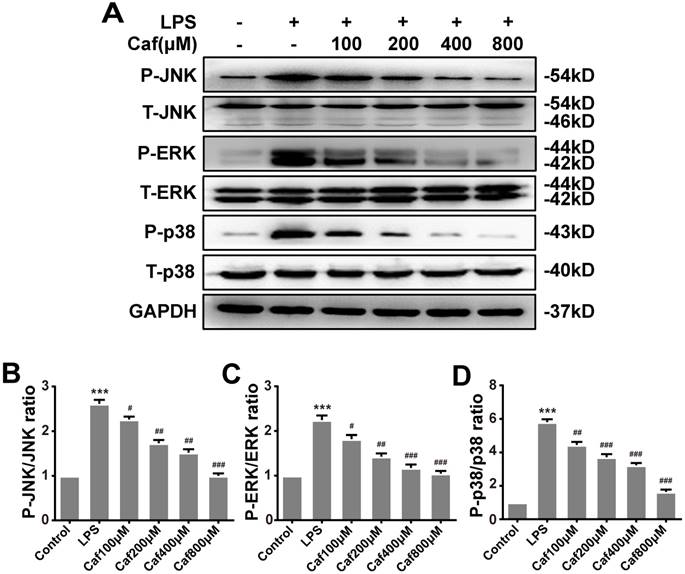
Emerging experimental evidence has indicated that both MAPK and NF-κB play important roles in the responses to inflammation stress and NLRP3 activation [31-33]. To investigate whether caffeine suppressed NLRP3 inflammasome activation by inhibiting MAPK/NF-κB signaling, we analyzed the protein levels and nuclear localization of NF-κB pathway molecules. The western blot results showed that caffeine pretreatment decreased the levels of phosphorylated IκB-α and p65 upon LPS stimulation (Figure 4A-C). We also found that pretreatment with caffeine significantly inhibited the nuclear localization of p65, implying its blocked activity (Figure 4D). Likewise, we detected decreased phosphorylation levels of JNK, ERK and p38 in cells stimulated by LPS following pretreatment with caffeine (Figure 5). The above results suggest that caffeine inhibits LPS-induced NLRP3 inflammasome activation by inhibiting MAPK/NF-κB signaling in THP-1 macrophages.
Caffeine inhibits NF-κB activation in THP-1 macrophages stimulated by LPS. (A) The protein levels of IκB-α, p65 and their phosphorylated forms (P-IκB-α and P-p65) were analyzed by western blotting in THP-1 macrophages pretreated with caffeine for 1 h and then incubated with LPS (1 μg/ml) for 3 h. (B) Densitometric analysis was used to quantify the phosphorylation of IκB-α at Ser32. (C) Densitometric analysis was used to quantify the phosphorylation of p65 at Ser536. The results represent the mean ± SD for three experiments. *** p< 0.001 vs. the control group. # p<0.05, ## p<0.01, and ### p< 0.001 vs. the LPS group. (D) p65 nuclear localization was visualized by immunofluorescence analysis with an anti-p65 (green) antibody in THP-1 macrophages pretreated with caffeine (800 μM) for 1 h followed by LPS (1 μg/ml) for 3 h. DAPI (blue) was used as a nuclear marker. Scale bar: 50 μm.
Caffeine suppresses MAPK pathway activation in THP-1 macrophages stimulated by LPS. (A) The protein levels of JNK, ERK, p38, and their phosphorylated forms (P-JNK, P-ERK, and P-p38) were evaluated by western blotting in THP-1 macrophages pretreated with caffeine for 1 h and then incubated with LPS (1 μg/ml) for 3 h. (B) Densitometric analysis was used to quantify the phosphorylation of JNK at Thr183/Tyr185. (C) Densitometric analysis was used to quantify the phosphorylation of ERK1/2 at Thr202/Tyr1204. (D) Densitometric analysis was used to quantify the phosphorylation of p38 at Thr180/Tyr182. The results represent the mean ± SD for three experiments. *** p< 0.001 vs. the control group. # p<0.05, ## p<0.01, and ### p< 0.001 vs. the LPS group.
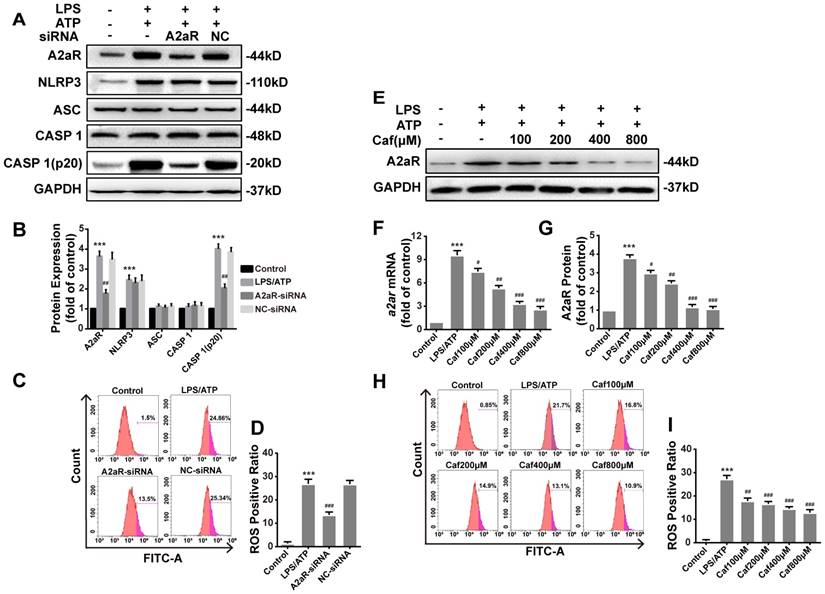
Caffeine inhibits NLRP3 inflammasome activation by suppressing A2aR-induced ROS production
Recent studies have shown that xanthines, including caffeine, exert their anti-inflammatory effects mainly by antagonizing A2aR [34] to reduce the production of ROS [35], which are required for activation of the NLRP3 inflammasome [36]. We first silenced A2aR expression by siRNA transfection of LPS and ATP stimulated THP-1 macrophages. After silencing, the protein levels of A2aR were significantly decreased, and notable downregulation of Caspase 1 (p20) expression was observed (Figure 6A and B). The expression of NLRP3, ASC and Caspase 1 was only slightly different in each treatment group compared with the negative control (NC) group (Figure 6A and B). In addition, ROS production in the A2aR siRNA group was lower than that in the NC group (Figure 6C and D). Then, we investigated the interaction between caffeine and A2aR in THP-1 macrophages. The results indicated that caffeine inhibited the expression of A2aR in a dose-dependent manner (Figure 6 E-G) and also inhibited ROS generation (Figure 6H and I). In summary, these findings indicate that caffeine inhibits ROS-mediated NLRP3 inflammasome activation at least partially by decreasing A2aR expression.
Caffeine inhibits A2aR activity to reduce the generation of ROS required for NLRP3 activation in THP-1 macrophages. (A) The protein levels of A2aR, NLRP3, ASC, Caspase 1 and Caspase 1 (p20) were evaluated by western blotting in THP-1 macrophages preincubated with A2aR-siRNA or negative control (NC)-siRNA for 12 h and then treated with LPS (1 μg/ml) for 3 h and ATP (5 mM) for 30 min. (B) Densitometric analysis was used to quantify the levels of A2aR, NLRP3, ASC, Caspase 1 and Caspase 1 (p20). (C) THP-1 macrophages were preincubated with A2aR-siRNA or NC-siRNA for 12 h and then treated with LPS (1 μg/ml) for 3 h and ATP (5 mM) for 30 min. ROS production was detected by flow cytometry. (D) Densitometric analysis was used to quantify the levels of ROS. (E) The protein levels of A2aR were evaluated by western blotting in THP-1 macrophages pretreated with caffeine for 1 h and then treated with LPS (1 μg/ml) for 3 h and ATP (5 mM) for 30 min. (F) The mRNA levels of a2ar were detected by qRT-PCR. (G) Densitometric analysis was used to quantify the levels of A2aR protein expression. (H) THP-1 macrophages were preincubated with caffeine for 1 h and then treated with LPS (1 μg/ml) for 3 h and ATP (5 mM) for 30 min. ROS production was detected by flow cytometry. (I) Densitometric analysis was used to quantify the levels of ROS. The results represent the mean ± SD for three experiments. *** p< 0.001 vs. the control group. # p<0.05, ## p<0.01, and ### p< 0.001 vs. the LPS and ATP group.
Discussion
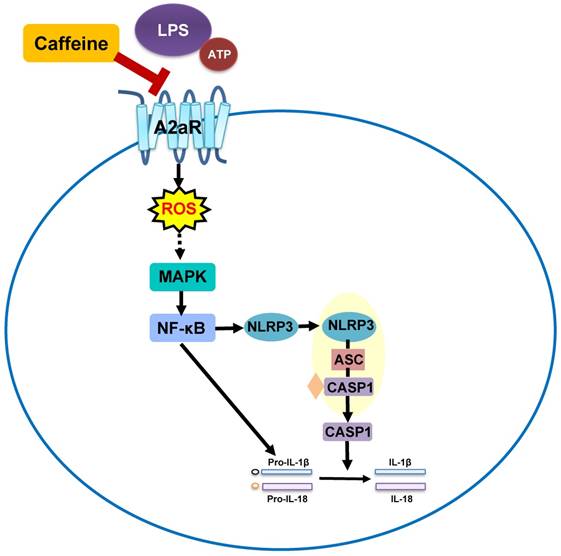
Notably, extensive studies on the lung-protective effects of caffeine in BPD have been conducted, but whether caffeine can alleviate NLRP3 inflammasome changes in BPD has remained unknown. In the present study, NLRP3 inflammasome levels in LPS-induced THP-1 macrophages were significantly reduced after pretreatment with caffeine. Moreover, MAPK/NF-κB signaling was inhibited by caffeine. RNA silencing of A2aR resulted in effects on the NLRP3 pathway similar to those of caffeine. Our study indicated that caffeine could inhibit the NLRP3 inflammasome in THP-1 macrophages by affecting MAPK/NF-κB signaling, in part by inhibiting A2aR-activated ROS generation (Figure 7). We thus propose a new mechanism for the lung-protective effects of caffeine.
Schematic diagram showing the effect of caffeine on LPS-stimulated NLRP3 inflammasome activation in THP-1 macrophages.
To date, a growing number of in vivo and in vitro studies have indicated that alveolar macrophages can be activated by prenatal chorioamnionitis combined with postpartum mechanical ventilation, oxygen therapy and nosocomial infection to secrete proinflammatory cytokines, including IL-1β, and thus further accelerate the progression of BPD [37-39]. In addition, investigators have verified that the release of IL-1β by alveolar macrophages is tightly controlled by activation of the NLRP3 inflammasome [40, 41]. Recently, Liao et al. [23] found that NLRP3-/- mice do not exhibit an inflammatory response under hyperoxia treatment and maintain normal alveolarization. Moreover, ventilated preterm baboons also show activation of the NLRP3 inflammasome and increased IL-1β levels. However, the effects of regular caffeine treatment on the NLRP3 inflammasome in activated macrophages during BPD remain poorly understood. In our study, caffeine treatment of THP-1 macrophages followed by LPS/ATP stimulation downregulated NLRP3, Caspase 1 and IL-1β. Our findings deepen understanding of the anti-inflammatory mechanism of caffeine and emphasize the need for further in vivo study.
It is well known that activation of the NLRP3 inflammasome is tightly regulated at the transcriptional and posttranslational levels. Previous studies have shown that in macrophages, LPS stimulates MAPK and NF-κB pathway activation as the first signal to transcriptional upregulating of IL-1β and IL-18 [42-44]. For NF-κB pathway, IκB-α is phosphorylated and thus marked for degradation by ubiquitin-dependent proteasomes upon LPS stimulation, which allows p65 phosphorylation and disassembled from the heterodimer. NF-κB p65, a representative subunit of the NF-κB heterodimer, is commonly phosphorylated at the Ser536 position and then translocate to the nucleus to upregulate NLRP3 and IL-1β mRNA expression [45]. In our study, caffeine markedly reduced the phosphorylation of IκB-α to prevent its degradation in LPS-stimulated THP-1 macrophages. At the same time, phosphorylation of p65 was decreased, as was activation and translocation of NF-κB. The MAPK pathway also participates in the activation of the NF-κB pathway as well as the upregulation of IL-1β and IL-18 [46, 47]. Our results demonstrated that caffeine inhibited the phosphorylation of JNK, ERK, and p38 induced by LPS. Taken together, the above results indicate that pretreatment with caffeine inhibits the transcriptional upregulation of IL-1β by interfering with MAPK-mediated NF-κB activity.
Caffeine has been reported to function as a nonselective adenosine receptor antagonist with a relatively high affinity for A2aR [48]. A2aRs are G protein-coupled receptors whose activation stimulates a series of downstream pathways, including the PKA, CREB, and MAPK pathways [49]. Recent experiments have shown that treatment with A2aR antagonists either prevents or reverses the ability of inflammatory stimuli to activate human alveolar macrophages [31]. In addition, A2aR acts as a key regulator of ROS production and is required for ATP-induced sustained NLRP3 inflammasome activation [50, 51]. Our results showed that knockdown of A2aR significantly impaired NLRP3 inflammasome activation and ROS production in LPS/ATP-stimulated THP-1 macrophages. Moreover, caffeine significantly inhibited LPS/ATP-induced A2aR gene transcription and protein expression in THP-1 macrophages, indicating that A2aR inhibition could act as a mediator by which caffeine suppresses NLRP3 activation.
Conclusion
In summary, our findings reveal that caffeine exerts anti-inflammatory effects by inhibiting NLRP3 inflammasome activation in THP-1 macrophages. These findings provide new insights and support a comprehensive understanding of the molecular mechanisms underlying the effects of caffeine treatment in BPD.
Abbreviations
A2AR: Adenosine A2A receptor; ATP: adenosine triphosphate; ASC: Apoptosis associated speck like protein containing a CARD; BPD: bronchopulmonary dysplasia; CAF: caffeine; CREB: cAMP response element-binding protein; DAPI: 4',6-diamidino-2-phenylindole; ERK: extracellular-regulated protein kinase; FBS: fetal bovine serum; IκB: inhibitor of κB; IL: interleukin; JNK: c-Jun N-terminal kinase; LPS: lipopolysaccharide; MAPK: mitogen-activated protein kinase; NC: negative control; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; NLRP3: nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3; PMA: phorbol 12-myristate 13-acetate; PKA: protein kinase A; ROS: reactive oxygen species; RPMI: Roswell Park Memorial Institute; siRNA: small interfering RNA; SP-B: surfactant protein B.
Acknowledgements
This study was supported by Shanghai Municipal Science and Technology Commission (134119a0500) and Key Developing Subject Program from Shanghai Municipal Commission of Health and Family Planning (No.2016ZB0102).
Authors' contributions
Z.W.M, M.L and G.X.H conceived and designed the study. Z.W.M and M.L performed the experiments and analyzed the data. Z.W.M, M.L, C.C and G.X.H wrote and revised the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Steinhorn R, Davis JM, Göpel W. et al. Chronic Pulmonary Insufficiency of Prematurity: Developing Optimal Endpoints for Drug Development. J Pediatr. 2017;191:15-21
2. Owen LS, Manley BJ, Davis PG. et al. The evolution of modern respiratory care for preterm infants. Lancet. 2017;389(10079):1649-1659
3. Matta P, Sherrod SD, Marasco CC. et al. In Utero Exposure to Histological Chorioamnionitis Primes the Exometabolomic Profiles of Preterm CD4(+) T Lymphocytes. J Immunol. 2017;199(9):3074-3085
4. van Haaften T, Byrne R, Bonnet S. et al. Airway delivery of mesenchymal stem cells prevents arrested alveolar growth in neonatal lung injury in rats. Am J Respir Crit Care Med. 2009;180(11):1131-1142
5. Villar J, Cabrera N, Casula M. et al. Mechanical ventilation modulates Toll-like receptor signaling pathway in a sepsis-induced lung injury model. Intensive Care Med. 2010;36(6):1049-1057
6. Ryan RM, Ahmed Q, Lakshminrusimha S. Inflammatory mediators in the immunobiology of bronchopulmonary dysplasia. Clin Rev Allergy Immunol. 2008;34(2):174-190
7. Kalymbetova TV, Selvakumar B, Rodriguez-Castillo JA. et al. Resident alveolar macrophages are master regulators of arrested alveolarization in experimental bronchopulmonary dysplasia. J Pathol. 2018;245(2):153-159
8. Iyengar A, Davis JM. Drug therapy for the prevention and treatment of bronchopulmonary dysplasia. Front Pharmacol. 2015;6:12
9. Schmidt B, Roberts RS, Davis P. et al. Caffeine therapy for apnea of prematurity. N Engl J Med. 2006;354(20):2112-2121
10. Schmidt B, Roberts RS, Davis P. et al. Long-term effects of caffeine therapy for apnea of prematurity. N Engl J Med. 2007;357(19):1893-1902
11. Pacifici GM. Clinical pharmacology of theophylline in preterm infants: effects, metabolism and pharmacokinetics. Curr Pediatr Rev. 2014;10(4):297-303
12. Endesfelder S, Weichelt U, Strauss E. et al. Neuroprotection by Caffeine in Hyperoxia-Induced Neonatal Brain Injury. Int J Mol Sci. 2017:18 (1)
13. Fehrholz M, Hutten M, Kramer BW. et al. Amplification of steroid-mediated SP-B expression by physiological levels of caffeine. Am J Physiol Lung Cell Mol Physiol. 2014;306(1):L101-109
14. Köroğlu OA, MacFarlane PM, Balan KV. et al. Anti-inflammatory effect of caffeine is associated with improved lung function after lipopolysaccharide-induced amnionitis. Neonatology. 2014;106(3):235-240
15. Lodha A, Seshia M, McMillan DD. et al. Association of early caffeine administration and neonatal outcomes in very preterm neonates. JAMA Pediatr. 2015;169(1):33-38
16. Chou WC, Kao MC, Yue CT. et al. Caffeine Mitigates Lung Inflammation Induced by Ischemia-Reperfusion of Lower Limbs in Rats. Mediators Inflamm. 2015;2015:361638
17. Fukumoto J, Fukumoto I, Parthasarathy PT. et al. NLRP3 deletion protects from hyperoxia-induced acute lung injury. Am J Physiol Cell Physiol. 2013;305(2):C182-189
18. Wu J, Yan Z, Schwartz DE. et al. Activation of NLRP3 inflammasome in alveolar macrophages contributes to mechanical stretch-induced lung inflammation and injury. J Immunol. 2013;190(7):3590-3599
19. Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol. 2012;28:137-161
20. Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821-832
21. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21(7):677-687
22. Benetti E, Chiazza F, Patel NS. et al. The NLRP3 Inflammasome as a novel player of the intercellular crosstalk in metabolic disorders. Mediators Inflamm. 2013;2013:678627
23. Liao J, Kapadia VS, Brown LS. et al. The NLRP3 inflammasome is critically involved in the development of bronchopulmonary dysplasia. Nat Commun. 2015;6:8977
24. Stouch AN, McCoy AM, Greer RM. et al. IL-1beta and Inflammasome Activity Link Inflammation to Abnormal Fetal Airway Development. J Immunol. 2016;196(8):3411-3420
25. Ma L, Xu H, Zhu J. et al. Ras1(CA) overexpression in the posterior silk gland improves silk yield. Cell Res. 2011;21(6):934-943
26. Ma L, Li K, Guo Y. et al. Ras-Raf-MAPK signaling promotes nuclear localization of FOXA transcription factor SGF1 via Ser91 phosphorylation. Biochim Biophys Acta Mol Cell Res. 2018;1865(4):560-571
27. Daly JW. Mechanism of action of caffeine. In: (ed.) Garattini S. Caffeine, coffee and health. New York: Raven Press. 1993:97-150
28. Beilharz M, De Nardo D, Latz E. et al. Measuring NLR Oligomerization II: Detection of ASC Speck Formation by Confocal Microscopy and Immunofluorescence. Methods Mol Biol. 2016;1417:145-158
29. Bauernfeind FG, Horvath G, Stutz A. et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183(2):787-791
30. Huang MT, Taxman DJ, Holley-Guthrie EA. et al. Critical role of apoptotic speck protein containing a caspase recruitment domain (ASC) and NLRP3 in causing necrosis and ASC speck formation induced by Porphyromonas gingivalis in human cells. J Immunol. 2009;182(4):2395-2404
31. Chen X, Wang N, Zhu Y. et al. The Antimalarial Chloroquine Suppresses LPS-Induced NLRP3 Inflammasome Activation and Confers Protection against Murine Endotoxic Shock. Mediators Inflamm. 2017;2017:6543237
32. Gan P, Gao Z, Zhao X. et al. Surfactin inducing mitochondria-dependent ROS to activate MAPKs, NF-kappaB and inflammasomes in macrophages for adjuvant activity. Sci Rep. 2016;6:39303
33. Moon JS, Lee S, Park MA. et al. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J Clin Invest. 2015;125(2):665-680
34. Boia R, Elvas F, Madeira MH. et al. Treatment with A2A receptor antagonist KW6002 and caffeine intake regulate microglia reactivity and protect retina against transient ischemic damage. Cell Death Dis. 2017;8(10):e3065
35. Li YF, Ouyang SH, Tu LF. et al. Caffeine Protects Skin from Oxidative Stress-Induced Senescence through the Activation of Autophagy. Theranostics. 2018;8(20):5713-5730
36. Zhou R, Yazdi AS, Menu P. et al. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221-225
37. Shrestha AK, Bettini ML, Menon RT. et al. Consequences of early postnatal lipopolysaccharide exposure on developing lungs in mice. Am J Physiol Lung Cell Mol Physiol. 2019;316(1):L229-l244
38. Kalymbetova TV, Selvakumar B, Rodriguez-Castillo JA. et al. Resident alveolar macrophages are master regulators of arrested alveolarization in experimental bronchopulmonary dysplasia. J Pathol. 2018;245(2):153-159
39. Willis GR, Fernandez-Gonzalez A, Anastas J. et al. Mesenchymal Stromal Cell Exosomes Ameliorate Experimental Bronchopulmonary Dysplasia and Restore Lung Function through Macrophage Immunomodulation. Am J Respir Crit Care Med. 2018;197(1):104-116
40. Wu J, Yan Z, Schwartz DE. et al. Activation of NLRP3 inflammasome in alveolar macrophages contributes to mechanical stretch-induced lung inflammation and injury. J Immunol. 2013;190(7):3590-3599
41. Fukumoto J, Fukumoto I, Parthasarathy PT. et al. NLRP3 deletion protects from hyperoxia-induced acute lung injury. Am J Physiol Cell Physiol. 2013;305(2):C182-189
42. Guha M, Mackman N. LPS induction of gene expression in human monocytes. Cell Signal. 2001;13(2):85-94
43. Boaru SG, Borkham-Kamphorst E, Van de Leur E. et al. NLRP3 inflammasome expression is driven by NF-kappaB in cultured hepatocytes. Biochem Biophys Res Commun. 2015;458(3):700-706
44. Oeckinghaus A, Hayden M S, Ghosh S. Crosstalk in NF-kappaB signaling pathways. Nat Immunol. 2011;12(8):695-708
45. Wu S, Li H, Yu L. et al. IL-1beta upregulates Muc5ac expression via NF-kappaB-induced HIF-1alpha in asthma. Immunol Lett. 2017;192:20-26
46. Meng Y, Yu C H, Li W. et al. Angiotensin-converting enzyme 2/angiotensin-(1-7)/Mas axis protects against lung fibrosis by inhibiting the MAPK/NF-kappaB pathway. Am J Respir Cell Mol Biol. 2014;50(4):723-736
47. Khan S, Choi R J, Shehzad O. et al. Molecular mechanism of capillarisin-mediated inhibition of MyD88/TIRAP inflammatory signaling in in vitro and in vivo experimental models. J Ethnopharmacol. 2013;145(2):626-637
48. Klinger M, Freissmuth M, Nanoff C, Adenosine receptors. G protein-mediated signalling and the role of accessory proteins. Cell Signal. 2002;14(2):99-108
49. Alfaro TM, Rodrigues DI, Tome AR. et al. Adenosine A2A receptors are up-regulated and control the activation of human alveolar macrophages. Pulm Pharmacol Ther. 2017;45:90-94
50. Ouyang X, Ghani A, Malik A. et al. Adenosine is required for sustained inflammasome activation via the A₂A receptor and the HIF-1α pathway. Nature communications. 2013;4:2909-2919
51. Thakur S, Du J, Hourani S. et al. Inactivation of adenosine A2A receptor attenuates basal and angiotensin II-induced ROS production by Nox2 in endothelial cells. J Biol Chem. 2010;285(51):40104-40113
Author contact
![]() Corresponding author: Xiaohui Gong, E-mail: gongxhcom.cn.
Corresponding author: Xiaohui Gong, E-mail: gongxhcom.cn.