Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Material and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(16):5074-5088. doi:10.7150/ijbs.84472 This issue Cite
Research Paper
S100a16 Deficiency Prevents Alcohol-induced Fatty Liver Injury via Inducing MANF Expression in Mice
Dan Wang1#, Rihua Zhang1#, Xiaoxuan Qin2, Jizheng Wang1, Yifang Hu1, Shan Lu1, Jingbao Kan1, Yaoqi Ge1, Ke Jin3 ![]() , Wen-Song Zhang1,4
, Wen-Song Zhang1,4 ![]() , Yun Liu1,5
, Yun Liu1,5 ![]()
1. Department of Geriatrics, The First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, 210029, China.
2. Department of Neurology, The First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, 210029, China.
3. Department of Infectious Diseases, The First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, 210029, China.
4. Department of Pharmacy, The First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, 210029, China.
5. Department of Medical Informatics, School of Biomedical Engineering and Informatics, Nanjing Medical University, Nanjing, Jiangsu, China.
#These authors contributed equally to this work.
Received 2023-3-20; Accepted 2023-8-26; Published 2023-10-2
Abstract

Alcoholic liver disease (ALD) encompasses conditions ranging from simple steatosis to cirrhosis and even liver cancer. It has gained significant global attention in recent years. Despite this, effective pharmacological treatments for ALD remain elusive, and the core mechanisms underlying the disease are not yet fully comprehended. S100A16, a newly identified calcium-binding protein, is linked to lipid metabolism. Our research has discovered elevated levels of the S100A16 protein in both serum and liver tissue of ALD patients. A similar surge in hepatic S100A16 expression was noted in a Gao-binge alcohol feeding mouse model. S100a16 knockdown alleviated ethanol-induced liver injury, steatosis and inflammation. Conversely, S100a16 transgenic mice showed aggravating phenomenon. Mechanistically, we identify mesencephalic astrocyte-derived neurotrophic factor (MANF) as a regulated entity downstream of S100a16 deletion. MANF inhibited ER-stress signal transduction induced by alcohol stimulation. Meanwhile, MANF silencing suppressed the inhibition effect of S100a16 knockout on ethanol-induced lipid droplets accumulation in primary hepatocytes. Our data suggested that S100a16 deletion protects mice against alcoholic liver lipid accumulation and inflammation dependent on upregulating MANF and inhibiting ER stress. This offers a potential therapeutic avenue for ALD treatment.
Keywords: Alcoholic liver disease, S100A16, hepatic steatosis, MANF, ER stress.
Introduction
Alcohol-associated liver disease (ALD) has emerged as a significant global public health concern, exerting immense pressure on healthcare systems [1-3]. Alcoholic fatty liver, the initial stage of ALD, is marked by hepatic steatosis, a process which has been identified as reversible. However, persistent alcohol intake can escalate this to more severe conditions such as alcoholic steatohepatitis, fibrosis, cirrhosis, and even hepatocellular carcinoma [4]. Alarmingly, over 50% of cirrhosis-related mortalities globally are linked to alcohol abuse [3]. To date, the precise mechanisms of ALD and effective interventions are yet to be fully elucidated [5]. This highlights the pressing necessity to uncover the disease's underlying pathogenesis and pave the way for novel therapeutic strategies.
The S100 calcium-binding protein 16 (S100A16), a recently identified member of the EF-hand S100 family proteins, exhibits broad expression across diverse human tissues, notably in the liver and adipose tissue [6]. In previous studies, we pinpointed S100A16 as a novel factor that promotes lipogenesis in the livers of high fat diet (HFD)-fed mice [7]. Additionally, the knockdown of S100a16 has shown potential in mitigating fatty liver injury. Our data further indicated that silencing of S100a16 can protect mice from liver fibrosis when exposed to stimuli such as CCl4, bile duct ligation surgery, and methionine choline deficiency diet feeding [8]. Moreover, a recent investigation revealed the protective effects of S100a16 knockdown against lipid accumulation and inflammation in HK-2 cells under high glucose conditions [9]. These findings underscore the potential role of S100A16 in the regulation of hepatic lipid metabolism and liver injury. Yet, its role in ALD's progression remains uncharted.
Endoplasmic reticulum stress (ER stress) is a cellular defense mechanism triggered in response to the accumulation of misfolded and unfolded proteins within the endoplasmic reticulum's lumen. This protective process initiates the unfolded protein responses (UPR). Three canonical UPR signaling pathways have been delineated: inositol-requiring protein-1 (IRE1), activating transcription factor-6 (ATF6), and protein kinase RNA (PKR)-like ER kinase (PERK) [10, 11]. Notably, the pathological link between ER stress and ALD has been substantiated through both experimental and clinical studies [12-15].
Mesencephalic astrocyte-derived neurotrophic factor (MANF) is an emerging dopaminergic neurotrophic factor believed to have regulating potential for ER stress [16, 17]. It has been confirmed that MANF is also a key regulator in hepatic lipid metabolism [18]. Hepatocyte-specific Manf overexpression could directly promote browning of white adipocytes via the p38 MAPK pathway, thereby counteracting HFD-induced obesity [19]. In addition, mice hepatocyte-specific Manf overexpression reduced lipid accumulation in liver, whereas Manf ablation exacerbated hepatic steatosis. Mechanistic insights revealed that MANF inhibition upregulated G0S2, a critical determinant that fostered lipid accumulation and suppressed lipolysis [19, 20].
In our current research, we observed a marked upregulation of S100A16 in ALD patients. Additionally, S100a16 knockdown mitigated the severity of alcohol-induced fatty liver injury in mice. Mechanistically, the surge in S100A16 induced by alcohol was found to inhibit MANF, which in turn fostered lipid accumulation and inflammatory reactions. Collectively, our findings posited S100A16 as a potential novel regulator in the pathogenesis of ALD.
Material and Methods
Human samples
Pathological section of liver and serum from subjects with ALD and healthy controls were obtained from the first affiliated hospital of Nanjing Medical University (Nanjing, China). All ALD patients had history of alcohol consumption averaging at least 70 g/day, for at least 20 years. We excluded patients with drug-induced liver injury, autoimmune liver disease, hepatitis B or C, and hepatocellular carcinoma. Samples from relative healthy subjects were known to have no history of excessive drinking or chronic liver disease. The information of ALD patients and healthy individuals are shown in Table 1. All research was conducted in accordance with both the Declarations of Helsinki and Istanbul. The study protocol and use of bio-samples for research were approved by the ethics committee of the First Affiliated Hospital of Nanjing Medical University (No.2022-SR-582).
Demographic and clinical parameters of healthy controls and patients with ALD.
| Variables | Healthy controls (n=9) | ALD patients (n=19) |
|---|---|---|
| Sex, male, n (%) | 88.9 | 89.5 |
| Age, y | 52.6±3.9 | 58.3±2.2 |
| ALT level, U/L | 19.2±0.8 | 170.7±49.9* |
| AST level, U/L | 21.5±0.7 | 150.7±29.2** |
| ALP level, U/L | 80.2±6.5 | 174±26.8* |
| GGT level, U/L | 23.5±2.1 | 232±50.5** |
| TBIL, μmol/L | 11.3±0.82 | 191.1±44.4** |
| Albumin level, g/dL | 45.4±0.6 | 31.4±1.2 |
| hepatic malignancies | Negative | Negative |
| Alcohol Consumption, g/d | Negative | ≥70 |
All the data are shown as mean ± SEM. * p < 0.05 vs. Control group. ** p < 0.01 vs. Control group.
Animal experiments
S100a16 knockout heterozygote (S100a16KO+/-) mice and S100a16 transgenic (S100a16TG) mice were generated as described previously [7, 8]. Briefly, the S100a16 allele with floxed exon 2-3 was backcrossed into the C57BL/6 background. It was then bred with EIIa-Cre mice expressing the Cre recombinase to generate S100a16KO+/- in over 98% C57BL/6 background. During this process, we found that S100a16 homozygous (S100a16KO-/-) mutations were lethal. So, we used S100a16 knockout heterozygote (S100a16KO+/-) mice in this research.
S100a16 mice cDNA was cloned into the pCAGGS plasmid, transgenic mice (S100a16TG mice) were generated by purified DNA fragments microinjection into the pronuclei of fertilized eggs of C57BL/6J.
Hepatocyte-specific S100a16 knockout (S100a16LKO) mice were generated by crossing S100a16-floxed mice with Albumin-Cre transgenic mice (Alb-Cre) with both strains on the C57BL/6J background. S100a16-floxed (S100a16f/f) mice were mated with Alb-Cre mice to generate S100a16LKO heterozygote mice. Then, S100a16LKO mice and their littermate controls (S100a16f/f mice) were obtained by mating S100a16LKO heterozygote mice with S100a16f/f mice.
All mice used in our experiments were identified by PCR analyses of the genomic DNA obtained from tail tips. All animal were housed in ventilated cages at the SPF facility in a temperature-controlled environment with a 12-hour light/dark cycle and had free access to water and diet. All animal experiments were approved by the Institutional Animal Care and Use Committee of Nanjing Medical University (No. IACUC-2105059).
Ethanol feeding studies
Age-matched eight- to ten-week-old male mice were treated with either an isocaloric ethanol or pair-fed control diet following the protocol of NIAAA model [21]. Briefly, all mice were first fed ad libitum with the control Lieber-DeCarli liquid diet (Bio-Serv, product No. F1259SP) for 5 days, Ethanol groups were then fed with a liquid diet (Bio-Serv, product No. F1258SP) containing 5% (vol/vol) ethanol for 10 days and control groups were fed with isocaloric control diet for 10 days. At day 11, mice were administrated by oral gavage in the morning with a single bolus of ethanol (5 g/kg body weight) or isocaloric maltose dextrin solution (9 g/kg body weight), respectively. The mice were sacrificed and blood and tissue samples were collected 9 h post-gavage. Liquid diets were freshly prepared from powder daily. Food intake was recorded daily. Depilation, refusal to eat, intolerable pain, and dramatic weight loss were used as humane endpoints in this experiment.
Serum and liver triglyceride (TG), total cholesterol (T-CHO) and serum alanine transaminase (ALT), aspartate transaminase (AST) levels were determined using biochemical assay kits (Nanjing Jiancheng Bioengineering Institute, A110-1-1, A111-1-1, C009-2-1 and C010-2-1) according to the manufacturer's specifications.
Cell culture and treatment
Mice primary hepatocytes were isolated by in situ perfusion digestion of livers using type IV collagenase (Gibco, 17104019), followed by separation using PERCOLL (sigma, P1644) and centrifugation. Isolated primary hepatocytes were maintained in DMEM medium supplemented with 10 % Fetal Bovine Serum (FBS, Gibco, 10270-106) and 1 % penicillin/streptomycin (Gibco, 15140122). The culture medium was replaced daily.
AML12 hepatocytes, a non-transformed mouse hepatocyte cell line, was maintained in DMEM/F12 medium (Gibco, 11330-032) supplemented with ITS liquid media supplement (Sigma, I3146), 40 ng/mL dexamethasone (Sigma, D4902-100mg) and 10 % FBS (Gibco, 10270-106). Cells were treated with medium containing 50 mM ethanol for 24 h and changed intervals fresh medium at 12 h. All the cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2.
Gene silencing and overexpression
For the knockdown and overexpression of S100a16 and Manf, the AML12 cell line was transfected with siRNA or a full-length plasmid using Lipofectamine® 2000 transfection reagent (Invitrogen, 11668019) according to the manufacturer's protocol. Knockdown and overexpression of Manf in S100a16LKO or S100a16TG mice primary hepatocytes were performed as above described. Negative scrambled control siRNA and empty vector plasmid (pcDNA3.1) were transfected into the control groups.
Protein isolation and Western blotting
Total protein was extracted from liver tissues and cells using RIPA lysis buffer (Beyotime P0013) added with protease inhibitors (biosharp BL612A) and phosphatase inhibitors (biosharp BL615A). Protein samples were mixed with loading buffer (Beyotime P0298) and separated by 6-12.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Then, the proteins were transferred onto nitrocellulose membranes (BioTraceTM NT T33517). Nitrocellulose membranes were blocked in TBST containing 5% skim milk for 2 h at room temperature. Then the membranes were incubated in primary antibodies overnight at 4 ℃, followed by incubation in the corresponding secondary antibodies. The relative expression levels of target proteins were quantified using ImageJ software (version 1.48). All primary antibodies used are listed in Table S1.
RNA isolation and RT-qPCR analysis
Total mRNA was extracted using RNA easy Isolation Reagent (Vazyme, R701-01). The concentration of RNA was quantified by a Nanodrop2000 spectrophotometer (Thermo Scientific, USA). cDNA was synthesized using HiScript III RT SuperMix for qPCR (Vazyme, R323-01) and detected using QuantStudio 7 system (Applied Biosystems). Relative mRNA expression levels were calculated using 2-ΔΔct method and were normalized against the levels of 18s. The primer sequences used for each gene are listed in Table S2.
Histological Examination (HE) staining
Fresh liver tissue was fixed in 4 % formaldehyde solution. Fixed liver tissues were embedded in paraffin, cut into 5 μm thick slices and stained with hematoxylin and eosin (H&E) following the standard protocol for analysis.
Oil Red O staining
Fresh liver tissue was embedded with OCT freeze embedding agent and frozen, and then cut into 5 μm thick slices. Oil-red was stained following the standard protocol for analysis. Slices were scanned by an automatic digital slide scanner.
BODIPY staining
Primary hepatocytes and AML 12 cells were first fixed with 4% paraformaldehyde followed by incubation with BODIPY® 493/503 (Invitrogen 2295013) for lipid staining. Cell nuclei were counterstained with DAPI (SouthernBiotech, H0621-V341). The lipid droplets were visualized under a THUNDER Imaging Systems (Leica, THUNDER DMi8).
Immunofluorescence
Liver tissue slices were permeabilized with 0.5 % Triton X-100 for 30 min (after deparaffinization) and blocked with 5 % goat serum for 1 h at RT. Then the slices were incubated with anti-S100A16 or anti-MANF primary antibodies overnight at 4°C, followed by an appropriate fluorescently tagged secondary antibody in the dark at RT for 1.5 h. Primary and secondary antibodies were diluted in 3 % BSA. DAPI was used to stain nuclei. Fluorescence microscopy was performed using a THUNDER Imaging Systems (Leica, THUNDER DMi8).
ELISA
Commercially available human ELISA kits (CUSABIO, Wuhan, China) were used to detect S100A16 levels in human serum (ALD and healthy individuals) according to the instructions provided by the manufacturer.
RNA sequencing and analysis
Primary hepatocytes (n=3/group) were isolated from alcohol-fed WT and S100a16KO+/- mice. Total RNA was extracted, and mRNA was purified. mRNA was fragmented and reverse transcribed to generate cDNA. Sequencing analysis was performed as our previous work [8]. Genes with p value < 0.05 and fold change ≥ 2 were considered differentially expressed using DESeq2 software between S100a16 silencing group and WT group. Gene functional annotations were assessed based on Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) databases. The raw sequence data have been submitted to the NCBI Gene Expression Omnibus (GEO) database (accession no. GSE243683).
Statistical analysis
All data are expressed as the mean ± SEM. Two-tailed student's t-test was used to calculate the statistical significance between two groups and one-way analysis of variance (one way ANOVA) for three or more groups. In all cases, p < 0.05 was considered statistically significant.
Results
Alcohol consumption up-regulated S100A16 expression in human and mice
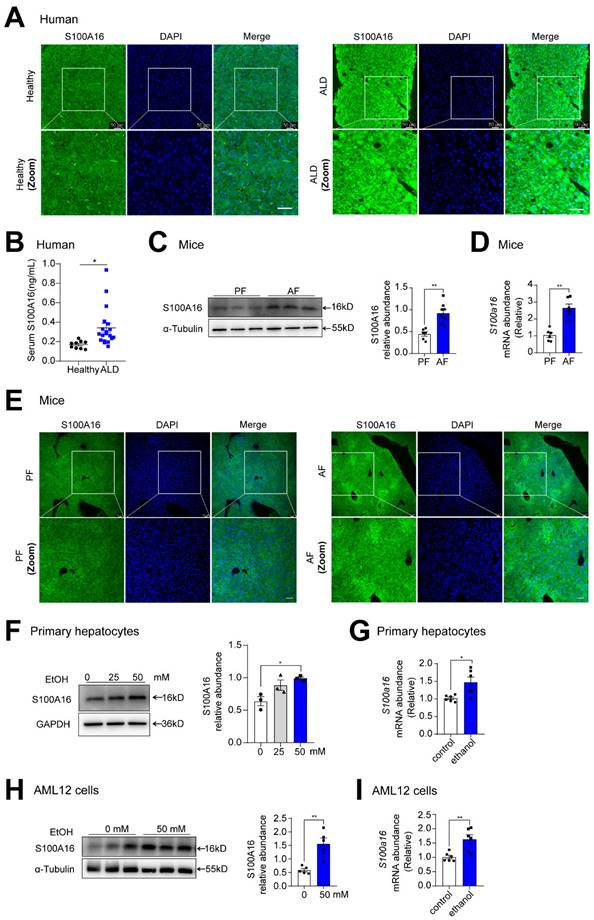
To investigate the potential involvement of S100A16 in ALD progression, we first evaluated S100A16 expression levels in patients with ALD and healthy controls. Both immunofluorescence and immunohistochemical assays indicated S100A16 was significantly increased in the ALD liver tissue compared with healthy individuals (Fig. 1A, S1A). Concurrently, serum S100A16 levels were found to be elevated in individuals diagnosed with ALD (Fig. 1B). Subsequently, we employed a short-term chronic plus one binge ethanol feeding model (NIAAA model). Analyses using western blot, RT-qPCR, and immunofluorescence staining collectively highlighted the heightened expression of S100A16 in the livers of alcohol-fed WT mice (Fig. 1C-E). In parallel, in vitro experiments illustrated that ethanol exposure upregulated S100A16 expression in primary hepatocytes as well as AML12 cells (Fig. 1F-I). Overall, these results suggested that alcohol consumption significantly increased hepatic S100A16 expression.
Alcohol consumption up-regulated S100A16 expression in human and mice. (A) Representative immunofluorescence staining of S100A16 in liver paraffin slices from healthy controls or patients with alcoholic-associated liver disease (ALD). Scale bar = 50 μm. (B) ELISA analysis of S100A16 expression in serum of ALD patients(n=19) and healthy controls(n=9). (C-E) Mice were pair-fed or alcohol-fed for 10 d and administered a single binge of ethanol (NIAAA model). (C) Western blot analysis of S100A16 expression in mice liver. Quantification of western blots on the right. (D) S100a16 mRNA levels in mice liver. (E) Immunofluorescence assessment of S100A16 expression in mice liver. (F-G) Protein and mRNA expression of S100A16 in primary hepatocytes treated with ethanol (50 mM) for 24 h. (H-I) Protein and mRNA expression of S100A16 in AML 12 cells treated with ethanol (50 mM) for 24 h. α-Tubulin or GAPDH served as the loading control. All data are represented as the mean ± SEM values. *: p < 0.05 vs. the control group, **: p < 0.01 vs. the control group. ALD: alcohol-associated liver disease. PF: pair-fed group. AF: alcohol-fed group.
S100a16 deletion protected mice against alcohol-induced fatty liver injury
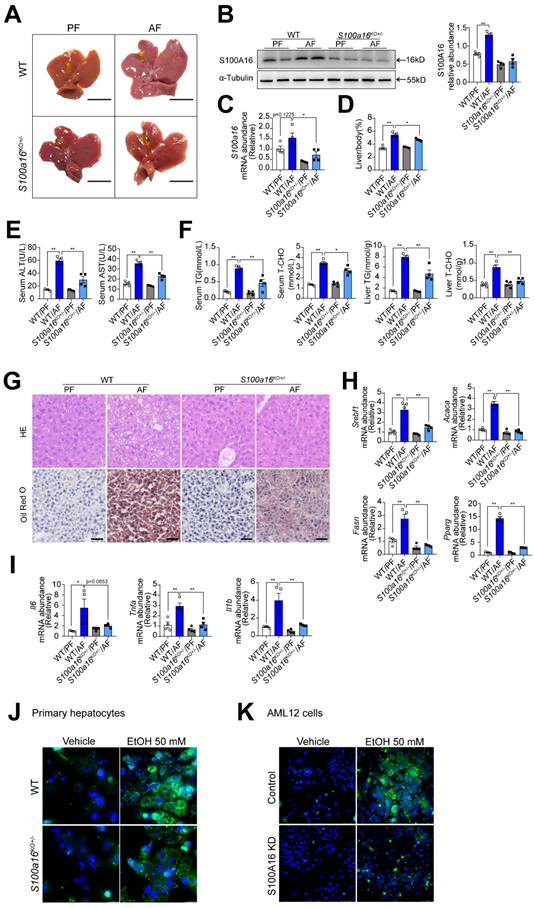
To elucidate the relationship between S100A16 and alcoholic fatty liver disease, we generated S100a16 knockout mice. Homozygous deletion of S100a16 in mice causes mortality, therefore we used S100a16 heterozygous knockout mice (S100a16KO+/- mice). Genotyping of S100a16KO+/- and wild type (WT) mice were performed before the experiment (Fig. S2A). Deletion of S100A16 in the liver of S100a16KO+/- mice was confirmed at the protein level (Fig. S2B). Our data showed that the livers became bigger in the WT group after ethanol exposure, but this condition was alleviated in alcohol-fed S100a16KO+/- mice (Fig. 2A). Western blot and RT-qPCR assays verified the augmented S100A16 expression in the livers of alcohol-fed WT mice (Fig. 2B-C). While body weights remained consistent between alcohol-fed S100a16KO+/- and WT mice (Fig. S2C), but liver/body weight ratios were mitigated in S100a16KO+/- group (Fig. 2D). As shown in Fig. 2E, knockout of S100a16 alleviated the upregulation of serum ALT, AST levels during alcohol consumption. Serum TG, TC contents and liver TG, TC contents all showed a decline in the S100a16KO+/- group (Fig. 2F). Liver histological studies, encompassing H&E and Oil Red O staining, pointed to a mitigated liver injury in the S100a16KO+/- cohort (Fig. 2G). Additionally, there was a marked suppression in the expression of crucial lipogenic and inflammatory genes in the livers of these mice upon genetic S100a16 inhibition (Fig. 2H-I). Also noteworthy, lipid droplets in ethanol-induced primary hepatocytes isolated from S100a16KO+/- mice showed a significant reduction when compared with that from WT mice (Fig. 2J). As shown in Fig. S2D, we employed siRNA to diminish S100A16 expression (S100A16 KD) in AML12 cells and the knockdown efficiency was confirmed using western blot. Analogous to our earlier findings, S100a16 knockdown led to diminished lipid droplets in AML12 cells (Fig. 2K). In summary, inhibiting S100A16 substantially alleviated the symptoms of alcohol-induced fatty liver injury.
S100a16 deletion protected mice against alcohol-induced fatty liver injury. WT or S100a16KO+/- mice were fed separately with the corresponding liquid diet 10 days and administered a single binge of ethanol. (A) Pictures of mice liver from different groups. Protein (B) and mRNA (C) levels of S100A16 in the liver from WT and S100a16KO+/- mice. Quantification of western blots on the right. (D) Liver weight/body weight (%). (E) Serum ALT, AST levels. (F) Serum TG, TC levels and liver TG, TC levels. (G) Representative H&E and Oil Red O staining of mice livers. Scale bars: 50 µm. (H) Relative mRNA levels of Srebf1, Acaca, Fasn, and Pparg. (I) Relative mRNA levels of Il6, Tnfa, and Il1b. (J) Primary hepatocytes from WT and S100a16KO+/-mice were treated with ethanol (50 mM) for 24 h. Representative images of BODIPY 493/503 staining of lipid droplets. Scale bars: 50 µm. (K) AML 12 cells with knockdown of S100A16 by siRNA (S100A16 KD) were treated with ethanol (50 mM) for 24 h. Representative images of BODIPY 493/503 staining of lipid droplets. Scale bars: 50 µm. α-Tubulin served as the loading control. All data are represented as the mean ± SEM values. *: p < 0.05 vs. the control group, **: p < 0.01 vs. the control group. ALT, alanine aminotransferase; AST, aspartate aminotransferase; TG: triacylglycerol; T-CHO: total cholesterol; H&E, hematoxylin and eosin.
S100a16 overexpression aggravated alcohol-induced fatty liver injury in mice
To further verify the relationship between S100A16 and alcoholic fatty liver injury diseases, S100a16 transgenic (S100a16TG) mice systemically overexpressing S100a16 were engineered under control of the pCAGGS vector promoter (Fig. S3A). Expression of S100A16 in the liver of S100a16TG mice was confirmed at the protein level (Fig. S3B). We then exposed age-matched WT and S100a16TG mice to the NIAAA model, observing the subsequent changes. Unlike the results from S100a16KO+/- mice, post-ethanol exposure, the livers of S100a16TG mice had larger livers than WT mice after ethanol exposure (Fig. 3A). Moreover, liver-to-body weight ratios surged in the S100a16TG group (Fig. 3D). Results showed that S100A16 overexpression in liver was increased in the alcohol-fed mice (Fig. 3B-C). Contrasting with the effects of S100a16 suppression, overexpression of S100a16 exacerbated the progression of alcohol-induced fatty liver development compared with WT mice. This exacerbation was evident both biochemically (assessing ALT, AST, TG, TC levels) and histologically (using H&E and Oil Red O staining) (Fig. 3E-G). The mRNA levels of molecules that promote fat accumulation and inflammation were also augmented in alcohol-exposed S100a16TG mice (Fig. 3H-I). Notably, ethanol-induced primary hepatocytes derived from S100a16TG mice exhibited a pronounced increase in lipid droplets compared with primary hepatocytes from WT mice (Fig. 3J). As shown in Fig. S3C, S100A16 overexpression efficiency in AML 12 cells was confirmed by western blot analysis. Similarly, overexpression of S100A16 in AML12 cells also amplified ethanol-induced lipid accumulation (Fig. 3K). Collectively, these findings compellingly argued that overexpressing S100A16 intensified the manifestations of alcohol-induced hepatic steatosis and injury.
S100a16 overexpression aggravated alcohol-induced fatty liver injury in mice. WT or S100a16TG mice were fed separately the corresponding liquid diet 10 days and administered a single binge of ethanol. (A) Pictures of mice liver from different groups. Protein (B) and mRNA (C) levels of S100A16 in the liver from WT and S100a16TG mice. Quantification of western blots on the right. (D) Liver weight/body weight (%). (E) Serum ALT, AST levels. (F) Serum TG, TC levels and liver TG, TC levels. (G) Representative H&E and Oil Red O staining of mice livers. Scale bars: 50 µm. (H) Relative mRNA levels of Srebf1, Acaca, Fasn, and Pparg. (I) Relative mRNA levels of Il6, Tnfa, and Il1b. (J) Primary hepatocytes from WT and S100a16TG mice were treated with ethanol (50 mM) for 24 h. Representative images of BODIPY 493/503 staining of lipid droplets. Scale bars: 50 µm. (K) AML 12 cells with overexpression of S100A16 by overexpression plasmid (S100A16+) were treated with ethanol (50 mM) for 24 h. Representative images of BODIPY 493/503 staining of lipid droplets. Scale bars: 50 µm. α-Tubulin served as the loading control. All data are represented as the mean ± SEM values. *: p < 0.05 vs. the control group, **: p < 0.01 vs. the control group.

S100a16 deficiency upregulated MANF expression in alcohol-fed mice
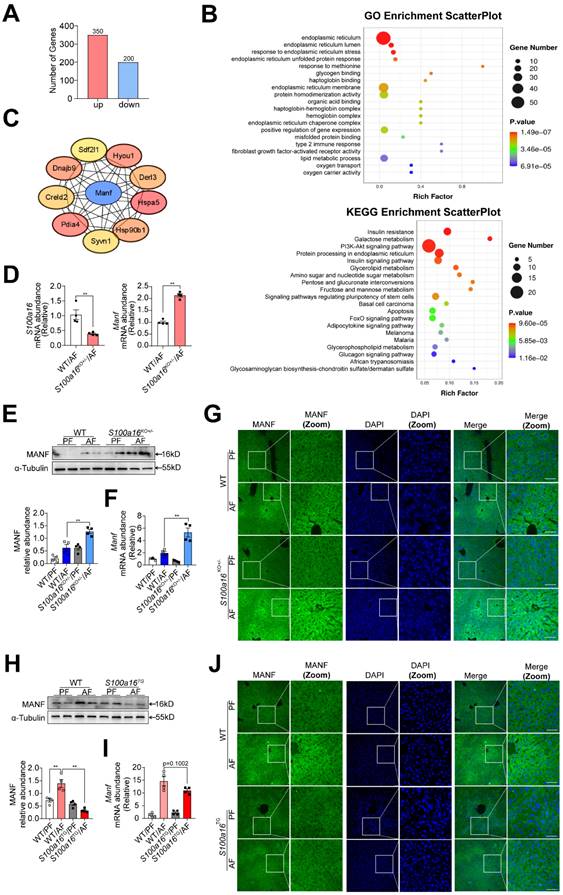
Subsequently, we examined the underlying mechanism of S100A16 in regulating alcoholic fatty liver injury. Primary hepatocytes were isolated from alcohol-fed WT or S100a16KO+/- mice subjected to NIAAA model and genome-wide analysis of gene expression was performed by transcriptome sequencing. Those detectable genes with a fold change ≥ 2 and p value < 0.05 were defined as differentially expressed genes, when compared with equivalents measured from WT cells. Consequently, S100a16KO+/- cells gave rise to 550 differentially expressed genes (350 upregulated and 200 downregulated, Fig. 4A). The PCA plot is presented in Fig. S4A. Utilizing GO and KEGG enrichment analyses, we discerned the distribution of these differentially expressed genes, uncovering that the absence of S100a16 significantly affected intracellular processes, including the endoplasmic reticulum stress (ER stress) pathway (Fig. 4B). We then constructed a protein-protein interaction network (Fig. S4B) and identified hub genes using STRING (https://stringdb.org/) and Cytoscape (version 3.8.0). Notably, among these hub genes (Fig. 4C), mesencephalic astrocyte-derived neurotrophic factor (MANF) has been implicated as pivotal in alcohol-induced liver diseases [22, 23]. Emerging studies have portrayed MANF as an ER-resident protein influencing various metabolic diseases, including alcoholic fatty liver [18, 19]. As shown in Fig. 4D, we verified the RNA sequencing result using RT-qPCR. In mice liver tissue, we also found that gene and protein expression of MANF were increased in WT mice after alcohol feeding and these alterations were further increased in S100a16KO+/- mice (Fig. 4E-G). Conversely, a decline in MANF gene and protein expression was observed in alcohol-fed S100a16TG mice (Fig. 4H-J). From these findings, we postulated that S100a16 suppression ameliorated alcoholic fatty liver injury by augmenting MANF expression.
S100a16 deficiency upregulated MANF expression in alcohol-fed mice. (A-C) Primary hepatocytes(n=3/group) were isolated from alcohol-fed WT and S100a16KO+/- mice and genome-wide analysis of gene expression was performed by transcriptome sequencing. (A) The number of up-regulation (350) and down-regulation(200) of differentially expressed genes (p ≤ 0.05, fold change ≥ 2). (B) GO and KEGG pathways enrichment analyses. (C) Hub genes of differentially expressed genes. We constructed a protein-protein interaction network and identified hub genes related to alcoholic fatty liver using STRING and Cytoscape. (D) mRNA levels of S100a16 and Manf in primary hepatocytes from WT and S100a16KO+/- mice after alcohol feeding. (E) protein and(F) mRNA levels of MANF in the alcohol-fed WT and S100a16KO+/- mice livers. Quantification of western blots on the right. (G)Representative immunofluorescence images of MANF in the alcohol-fed WT and S100a16KO+/- mice livers. Scale bar: 50 μm. (H) protein and(I) mRNA levels of MANF in the alcohol -fed WT or S100a16TG mice livers. Quantification of western blots on the right. (J) Representative immunofluorescence images of MANF in the alcohol -fed WT and S100a16TG mice livers. Scale bar:50 μm. α-Tubulin served as the loading control. All data are represented as the mean ± SEM values. *: p < 0.05 vs. the control group, **: p < 0.01 vs. the control group. MANF: mesencephalic astrocyte-derived neurotrophic factor.
To more accurately confirm the role of S100A16 in alcoholic fatty liver disease, S100a16f/f mice were crossed with Albumin-Cre mice to generate hepatocyte-specific S100A16 knockout mice (genotype: S100a16LKO) and control mice (genotype: S100a16f/f) (Fig. S5A-B). Deletion of S100A16 in the liver of S100a16LKO mice was confirmed at the protein level (Fig. S5C). mRNA and protein levels of MANF were increased in the livers from alcohol-fed S100a16LKO mice compare with S100a16f/f mice (Fig. S5D-E). Similar with S100a16KO+/- mice, liver/body weight ratios were alleviated in S100a16LKO group (Fig. S5F). As depicted in Fig. S5G-I, the specific knockout of S100A16 in hepatocytes attenuated alcohol-induced fatty liver development, as demonstrated by both biochemical assay (ALT, AST, TG, TC levels) and histological examination (H&E staining). We also observed that hepatocyte-specific genetic suppression of S100a16 significantly curtailed the expression of key lipogenic and inflammatory genes in mice livers (Fig. S5J-K).
S100A16 restrained MANF and activated ER stress in ALD
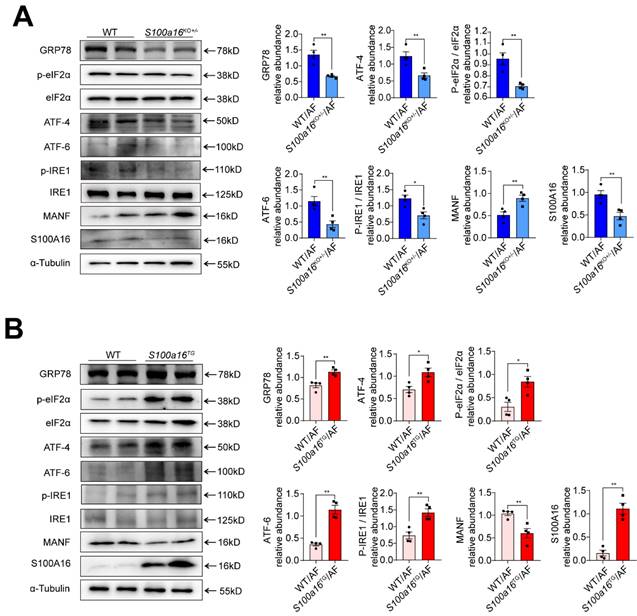
MANF, situated within the ER lumen, serves as a crucial shield against ER stress-induced cellular damage [24]. Given previous reports that ER stress is intricately linked with the pathogenesis of alcohol-induced fatty liver disease [15], we examined the expression of typical factors related to ER stress. Our findings revealed that, in alcohol-exposed S100a16KO+/-mice livers, there was a pronounced reduction in the expression of ER stress markers such as GRP78, ATF4, ATF6, and in the phosphorylation levels of IRE1α and EIF2α compared with their WT counterparts (Fig. 5A). Conversely, these trends were inverted in the S100a16TG mice (Fig. 5B). In summation, S100a16 ablation augmented MANF expression, thereby curtailing ER stress, which in turn mitigates the progression of alcoholic fatty liver development.
S100A16 restrained MANF and activated ER stress in ALD. (A) Western blot analysis of the S100A16, MANF and the ER stress-related genes GRP78, p-IRE1α, IRE1α, ATF6, ATF4, p-EIF2α, EIF2α in the livers of alcohol-fed WT or S100a16KO+/- mice. Quantification of western blots on the right. (B) Western blot analysis of the S100A16, MANF and the ER stress-related genes in the livers of alcohol-fed WT or S100a16TG mice. Quantification of western blots on the right. α-Tubulin served as the loading control. All data are represented as the mean ± SEM values. *: p < 0.05 vs. the control group, **: p < 0.01 vs. the control group. ATF4: activating transcription factor-4; ATF6: activating transcription factor-6; EIF2: eukaryotic initiation factor 2; GRP78: glucose-regulated protein 78; IRE1: inositol-requiring protein-1.
S100A16 regulated alcohol-induced lipid accumulation dependent on altering MANF expression
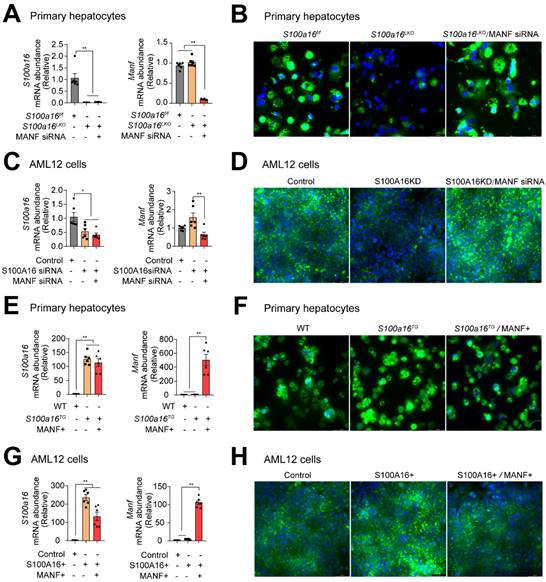
To ascertain whether S100A16 mediated ethanol-induced lipid accumulation dependent on MANF, we silenced MANF in primary hepatocytes isolated from S100a16LKO mice. RT-qPCR analyses authenticated the deletion of both S100A16 and MANF in primary hepatocytes (Fig. 6A). Observations from BODIPY staining indicated that Manf siRNA suppressed the inhibiting effects of S100a16 deletion on fat accumulation (Fig. 6B). Double knockdown of Manf and S100a16 in AML12 cells also confirmed these results (Fig. 6C-D). Elevated TG levels further substantiated that double knockdown of Manf and S100a16 in primary hepatocytes/AML12 cells inhibited the lipid-reducing impact of S100a16 deletion (Fig. S6A-B). Conversely, we induced overexpression of Manf in primary hepatocytes from S100a16TG mice, RT-qPCR analyses confirmed elevated levels of both S100a16 and Manf (Fig. 6E). Results showed that MANF overexpression significantly counteracted the lipid-enhancing effects of S100a16 overexpression (Fig. 6F). This observation was also corroborated in AML 12 cells (Fig. 6G-H). TG levels showed the same results (Fig. S6C-D). In essence, these outcomes suggested that the protective role of S100a16 deletion protected hepatocytes against alcohol-induced lipid accumulation dependent on promoting MANF expression.
S100A16 regulated alcohol-induced lipid accumulation dependent on altering MANF expression. (A) mRNA levels of S100a16 and Manf in ethanol-induced primary hepatocytes from S100a16f/f or S100a16LKO mice transfected with Manf siRNA. (B) Representative BODIPY staining images are shown on the right side, Scale bar: 50 μm. (C) mRNA levels of S100a16 and Manf in ethanol-induced AML 12 cells co-transfected with S100a16 and Manf siRNA. (D) Representative BODIPY staining images are shown on the right side, Scale bar: 50 μm. (E) mRNA levels of S100a16 and Manf in ethanol-induced primary hepatocytes from WT or S100a16TG mice transfected with overexpression plasmid. (F) Representative BODIPY staining images are shown on the right side, Scale bar: 50 μm. (G) mRNA levels of S100a16 and Manf in ethanol-induced AML 12 cells co-transfected with S100A16 and MANF overexpression plasmid. (H) Representative BODIPY staining images are shown on the right side, Scale bar: 50 μm. All data are represented as the mean ± SEM values. *: p < 0.05 vs. the control group, **: p < 0.01 vs. the control group. S100a16LKO: hepatocyte-specific S100a16 knockout.
Discussion
Alcohol abuse stands as a prime cause of liver-related mortalities globally. Despite its grave implications, there remains an absence of efficacious treatments beyond lifestyle adjustments [5]. This underscores the pressing need to identify innovative therapeutic targets for ALD. Our earlier investigations revealed that S100A16 modulated fatty liver development under high-fat diet conditions and high glucose-induced lipid accumulation [7, 9]. Both ALD and NAFLD are progressive conditions transitioning from steatosis to steatohepatitis, fibrosis, and eventually cirrhosis. While they share certain facets, distinct clinical characteristics and molecular mechanics demarcate them. For instance, ALD is more characterized by inflammatory cell infiltration, whereas lipid droplet accumulation is predominant in NAFLD. Despite these differences, existing interventions for both ailments are limited [25, 26]. Guided by our prior research, this study aimed to discern S100A16's role in ALD. Our findings revealed an up-regulation of S100A16 in both ALD patients and alcohol-exposed mice. S100A16 emerged as a catalyst for lipogenesis and inflammation, modulating MANF expression and ER stress signaling cascades (Fig. 7).
Schematic representation of the role of S100A16 in the pathogenesis of ALD. Alcohol consumption induced S100A16 upregulation. S100A16 aggravated ethanol-induced liver injury, steatosis and inflammation through inhibiting MANF expression and promoting ER stress-related signaling pathways.

S100 proteins traditionally influence myriad cellular functions, from differentiation and cycle progression to motility [27]. Recent findings have also unveiled their role in liver lipid metabolism. Specifically, S100A8 aggravated hepatitis in high fat and cholesterol-fed mice [28], while S100A11 spurs hepatic steatosis via the RAGE-mediated AKT-mTOR signaling axis [29]. Like most members of the S100 family, S100A16 was firstly reported as a regulatory factor related to malignant transformation and tumor development [6, 30-32]. Despite these known effects of S100A16, its role in other diseases is just emerging. Our previous studies showed the association of S100A16 with adipogenesis [33, 34], NFALD [7] and liver fibrosis [8]. Our current study underscored its upregulation in the liver and serum of ALD patients, suggesting its role in both ALD and NAFLD despite their different pathogenic mechanisms. These findings disclose an unrecognized mechanism for the role of S100A16 as a potential target for ALD and expose new insights into the action of other novel therapeutic targets for NAFLD to alleviate ALD progression.
Another major finding of this study is that we identified MANF, an endoplasmic reticulum-resident protein, as the downstream of S100A16 in alcoholic liver lipid accumulation. MANF is an emerging therapeutic target for metabolic diseases, including diabetes [35], obesity [19], fatty liver [20], alcohol-related diseases, and cardiovascular diseases [36]. Hepatic MANF overexpression improved adipose inflammation, insulin sensitivity and hepatic steatosis in HFD-fed mice [19]. Conversely, Hepatic MANF knockdown increased free fatty acid uptake and free fatty acid synthesis, which caused hepatic lipid accumulation and accelerated fatty liver development [20]. MANF deficiency also accelerated lipogenesis and exacerbated lipid accumulation in HepG2 cells [37]. In addition, MANF alleviated alcohol-induced hepatic steatosis via alleviating ER stress or activating STAT3-mediated autophagy [22, 23]. These events indicated that MANF is highly expressed in liver and plays an important role in the regulation of liver disease. In this study, transcriptomics results showed a significant upregulation of MANF in primary hepatocytes isolated from S100a16KO+/- mice compared with cells from WT. We further confirmed that MANF expression in liver was markedly promoted in alcohol-fed S100a16KO+/- mice. Reverse results were observed in alcohol-fed S100a16TG mice livers. Therefore, we speculated that S100A16 downregulated MANF expression and promoted alcohol-induced liver lipid accumulation and inflammation responses. In addition, MANF silencing abrogated the inhibitory effects of S100a16 knockout on lipid droplets formation in primary hepatocytes isolated from S100a16LKO mice in ethanol-containing medium. In contrast, MANF overexpression abrogated the protective effects of S100a16 transgene on lipid droplet formation. Similar results were confirmed in AML12 cells cultured medium containing ethanol. These results provided evidences that S100a16 deletion alleviates alcoholic fatty liver injury dependent on upregulating MANF expression. Besides, we also evaluated how S100A16 regulate MANF expression. We found S100A16 inhibited MANF expression using RNA sequencing, so we speculated that S100A16 transcriptionally regulated MANF. Indeed, S100A16 co-transfection decreased the luciferase activity of MANF promoter (Fig. S6E). A cycloheximide (CHX) chase assay was performed in S100A16 plasmid-transfected AML 12 cells to investigate the effect of S100A16 on the degradation rate of MANF protein. When protein synthesis was inhibited with CHX to exclude the potential effects, MANF protein was degraded in a time-dependent manner and S100A16 overexpression resulted in faster degradation, indicating that S100A16 promoted MANF degradation (Fig. S6F). Based on these results, we assumed that S100A16 inhibited MANF expression both by inhibiting its mRNA transcription and accelerating protein degradation process.
ER stress participated in the regulatory process of many diseases, including metabolic disorders [38], cancer [39] and neurological disorders [40]. It has been reported that S100 family proteins play a specific role in regulating ER stress. S100A1 expression is inhibited by CTCF and activates the PERK signaling pathway of ER stress in cardiomyocytes [41]. Hepatic steatosis and liver injury exacerbation of genetic mutations- or HFD-induced obesity is associated with an impaired UPR and persistent ER stress in the liver [42]. In our previous work about renal tubulointerstitial fibrosis, we found that the interaction between S100A16 and GRP78 is mediated through the IRE1α pathway and activated ER stress [43]. According to recent reports, ER stress upregulates NNMT through PERK-ATF4 pathway and promotes the development of ALD, indicating the role of ER stress in ALD pathogenesis [15]. Consistent with these reports, we also found that S100A16 influenced MANF expression and regulated ER stress to aggravate alcohol-induced lipid metabolism. In conclusion, our data provided evidence that S100A16 is a novel regulatory factor in ALD. S100A16 transcriptionally downregulated MANF and aggravated ER stress to mediate ALD responses. These findings suggested the translational potential of S100A16 as a therapeutic target for ALD.
It has been reported that males are more likely than females to consume alcohol and males are diagnosed with alcohol use disorders more often than females [44]. In addition, male and female have different reaction to alcohol consumption because of genetic patterns, hormone expression, and alcohol metabolism [45]. Further studies are needed in order to understand the differences between male and female in ALD.
In summation, our insights underscore S100A16 as a novel ALD regulatory factor, affecting MANF expression and exacerbating ER stress. This points to S100A16's potential as a promising therapeutic bullseye for ALD.
Abbreviations
AF: alcohol-fed group; ALD: alcohol-associated liver disease; ALT: alanine aminotransferase; AST: aspartate aminotransferase; ATF4: activating transcription factor-4; ATF6: activating transcription factor-6; EIF2: eukaryotic initiation factor 2; ER stress: endoplasmic reticulum stress; FBS: fetal bovine serum; GAPDH: glyceraldehyde-3-phosphatedehydrogenase; GRP78: glucose-regulated protein 78; HE: hematoxylin and eosin; IRE1: inositol-requiring protein-1; MANF: mesencephalic astrocyte-derived neurotrophic factor; PF: pair-fed group; RT-qPCR: quantitative real-time polymerase chain reaction; S100A16: S100 calcium binding protein 16; S100a16LKO: hepatocyte-specific S100a16 knockout; TG: triglyceride; T-CHO: total cholesterol; UPR: unfolded protein responses; WT: wild type.
Supplementary Material
Supplementary materials and methods, figures and tables.
Acknowledgements
We gratefully acknowledge associate Prof. Qun Liu from China Pharmaceutical University for kindly providing AML12 cells. We would like to thank the Core Facility of the First Affiliated Hospital of Nanjing Medical University for its help in the experiment.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 82000783), Natural Science Foundation of Jiangsu Province (BK20231366), the fellowship of China Postdoctoral Science Foundation (Grant Nos. 2020T130313 and 2019M661892), the National Key Research & Development Plan of the Ministry of Science and Technology of China (Grant Nos. 2018YFC1314900 and 2018YFC1314901), and the industry prospecting and common key technology projects of Jiangsu Province Science and Technology Department (Grant No. BE2020721).
Ethics approval and consent to participate
We obtained the informed consent before the study. Our study was approved by the Ethics Committee of the first Affiliated Hospital of Nanjing Medical University (No.2022-SR-582). Our animal research was approved by the Institutional Animal Care and Use Committee of Nanjing Medical University (No. IACUC-2105059).
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Author contributions
Dan Wang: Conceptualization, Data curation, Formal analysis, Resources, Investigation, Software, Visualization, Writing-original draft. Rihua Zhang: Data curation, Formal analysis, Methodology, Investigation, Writing-review & editing. Xiaoxuan Qin: Resources, Investigation, Software. Jizheng Wang: Resources, Investigation, Software. Yifang Hu: Resources, Investigation, Software. Shan Lu: Resources, Investigation, Software. Jingbao Kan: Investigation, Software. Yaoqi Ge: Resources, Software. Ke Jin: Conceptualization, Project administration, Writing-review & editing. Wen-Song Zhang: Conceptualization, Funding acquisition, Project administration, Writing-review & editing. Yun Liu: Conceptualization, Project administration, Supervision, Validation, Writing-review & editing.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Blachier M, Leleu H, Peck-Radosavljevic M, Valla DC, Roudot-Thoraval F. The burden of liver disease in Europe: a review of available epidemiological data. J Hepatol. 2013;58(3):593-608
2. Pimpin L, Cortez-Pinto H, Negro F, Corbould E, Lazarus JV, Webber L. et al. Burden of liver disease in Europe: Epidemiology and analysis of risk factors to identify prevention policies. J Hepatol. 2018;69(3):718-735
3. WHO. Global status report on alcohol and health 2014. 2014.
4. O'Shea RS, Dasarathy S, McCullough AJ; Practice Guideline Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Alcoholic liver disease. Hepatology. 2010;51(1):307-28
5. Louvet A, Mathurin P. Alcoholic liver disease: mechanisms of injury and targeted treatment. Nat Rev Gastroenterol Hepatol. 2015;12(4):231-42
6. Marenholz I, Heizmann CW. S100A16, a ubiquitously expressed EF-hand protein which is up-regulated in tumors. Biochem Biophys Res Commun. 2004;313(2):237-44
7. Kan J, Zhao C, Lu S, Shen G, Yang J, Tong P. et al. S100A16, a novel lipogenesis promoting factor in livers of mice and hepatocytes in vitro. J Cell Physiol. 2019;234(11):21395-21406
8. Zhang WS, Zhang R, Ge Y, Wang D, Hu Y, Qin X. et al. S100a16 deficiency prevents hepatic stellate cells activation and liver fibrosis via inhibiting CXCR4 expression. Metabolism. 2022;135:155271
9. Liu L, Lan S. Interference of S100A16 suppresses lipid accumulation and inflammation in high glucose-induced HK-2 cells. Int Urol Nephrol. 2021;53(6):1255-1263
10. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519-29
11. Cao SS, Kaufman RJ. Unfolded protein response. Curr Biol. 2012;22(16):R622-6
12. Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol. 2011;54(4):795-809
13. Longato L, Ripp K, Setshedi M, Dostalek M, Akhlaghi F, Branda M. et al. Insulin resistance, ceramide accumulation, and endoplasmic reticulum stress in human chronic alcohol-related liver disease. Oxid Med Cell Longev. 2012;2012:479348
14. Petrasek J, Iracheta-Vellve A, Csak T, Satishchandran A, Kodys K, Kurt-Jones EA. et al. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc Natl Acad Sci U S A. 2013;110(41):16544-9
15. Song Q, Chen Y, Wang J, Hao L, Huang C, Griffiths A. et al. ER stress-induced upregulation of NNMT contributes to alcohol-related fatty liver development. J Hepatol. 2020;73(4):783-793
16. Petrova P, Raibekas A, Pevsner J, Vigo N, Anafi M, Moore MK. et al. MANF: a new mesencephalic, astrocyte-derived neurotrophic factor with selectivity for dopaminergic neurons. J Mol Neurosci. 2003;20(2):173-88
17. Petrova PS, Raibekas A, Pevsner J, Vigo N, Anafi M, Moore MK. et al. Discovering novel phenotype-selective neurotrophic factors to treat neurodegenerative diseases. Prog Brain Res. 2004;146:168-83
18. Tang Q, Li Y, He J. MANF: an emerging therapeutic target for metabolic diseases. Trends Endocrinol Metab. 2022;33(4):236-246
19. Wu T, Liu Q, Li Y, Li H, Chen L, Yang X. et al. Feeding-induced hepatokine, Manf, ameliorates diet-induced obesity by promoting adipose browning via p38 MAPK pathway. J Exp Med. 2021;218(6):e20201203
20. Sousa-Victor P, Neves J, Cedron-Craft W, Ventura PB, Liao CY, Riley RR. et al. MANF regulates metabolic and immune homeostasis in ageing and protects against liver damage. Nat Metab. 2019;1(2):276-290
21. Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc. 2013;8(3):627-37
22. Chhetri G, Liang Y, Shao J, Han D, Yang Y, Hou C. et al. Role of mesencephalic astrocyte-derived neurotrophic factor in alcohol-induced liver injury. Oxid Med Cell Longev. 2020;2020:9034864
23. Zhang G, Liu Q, Li Y, Huang C, Zhou J, Zhao Y. et al. Mesencephalic astrocyte-derived neurotrophic factor alleviates alcohol induced hepatic steatosis via activating Stat3-mediated autophagy. Biochem Biophys Res Commun. 2021;550:197-203
24. Apostolou A, Shen Y, Liang Y, Luo J, Fang S. Armet, a UPR-upregulated protein, inhibits cell proliferation and ER stress-induced cell death. Exp Cell Res. 2008;314(13):2454-67
25. Tannapfel A, Denk H, Dienes HP, Langner C, Schirmacher P, Trauner M. et al. Histopathological diagnosis of non-alcoholic and alcoholic fatty liver disease. Virchows Arch. 2011;458(5):511-23
26. Zhang P, Wang W, Mao M, Gao R, Shi W, Li D. et al. Similarities and differences: A comparative review of the molecular mechanisms and effectors of NAFLD and AFLD. Front Physiol. 2021;12:710285
27. Heizmann CW, Fritz G, Schäfer BW. S100 proteins: structure, functions and pathology. Front Biosci. 2002;7:d1356-68
28. Mukai K, Miyagi T, Nishio K, Yokoyama Y, Yoshioka T, Saito Y. et al. S100A8 production in CXCR2-expressing CD11b+Gr-1high cells aggravates hepatitis in mice fed a high-fat and high-cholesterol diet. J Immunol. 2016;196(1):395-406
29. Teng F, Jiang J, Zhang J, Yuan Y, Li K, Zhou B. et al. The S100 calcium-binding protein A11 promotes hepatic steatosis through RAGE-mediated AKT-mTOR signaling. Metabolism. 2021;117:154725
30. Zhou W, Pan H, Xia T, Xue J, Cheng L, Fan P. et al. Up-regulation of S100A16 expression promotes epithelial-mesenchymal transition via Notch1 pathway in breast cancer. J Biomed Sci. 2014;21(1):97
31. Fang D, Zhang C, Xu P, Liu Y, Mo X, Sun Q. et al. S100A16 promotes metastasis and progression of pancreatic cancer through FGF19-mediated AKT and ERK1/2 pathways. Cell Biol Toxicol. 2021;37(4):555-571
32. Bresnick AR, Weber DJ, Zimmer DB. S100 proteins in cancer. Nat Rev Cancer. 2015;15(2):96-109
33. Liu Y, Zhang R, Xin J, Sun Y, Li J, Wei D. et al. Identification of S100A16 as a novel adipogenesis promoting factor in 3T3-L1 cells. Endocrinology. 2011;152(3):903-11
34. Li D, Zhang R, Zhu W, Xue Y, Zhang Y, Huang Q. et al. S100A16 inhibits osteogenesis but stimulates adipogenesis. Mol Biol Rep. 2013;40(5):3465-73
35. Fu J, Nchambi KM, Wu H, Luo X, An X, Liu D. Liraglutide protects pancreatic β cells from endoplasmic reticulum stress by upregulating MANF to promote autophagy turnover. Life Sci. 2020;252:117648
36. Arrieta A, Blackwood EA, Stauffer WT, Santo Domingo M, Bilal AS, Thuerauf DJ. et al. Mesencephalic astrocyte-derived neurotrophic factor is an ER-resident chaperone that protects against reductive stress in the heart. J Biol Chem. 2020;295(22):7566-7583
37. He M, Wang C, Long XH, Peng JJ, Liu DF, Yang GY. et al. Mesencephalic astrocyte-derived neurotrophic factor ameliorates steatosis in HepG2 cells by regulating hepatic lipid metabolism. World J Gastroenterol. 2020;26(10):1029-1041
38. Tirosh A, Tuncman G, Calay ES, Rathaus M, Ron I, Tirosh A. et al. Intercellular transmission of hepatic ER Stress in obesity disrupts systemic metabolism. Cell Metab. 2021;33(2):319-333.e6
39. Chen X, Cubillos-Ruiz JR. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat Rev Cancer. 2021;21(2):71-88
40. Uddin MS, Yu WS, Lim LW. Exploring ER stress response in cellular aging and neuroinflammation in Alzheimer's disease. Ageing Res Rev. 2021;70:101417
41. Zeng Z, Huang N, Zhang Y, Wang Y, Su Y, Zhang H. et al. CTCF inhibits endoplasmic reticulum stress and apoptosis in cardiomyocytes by upregulating RYR2 via inhibiting S100A1. Life Sci. 2020;242:117158
42. Bailly-Maitre B, Belgardt BF, Jordan SD, Coornaert B, von Freyend MJ, Kleinridders A. et al. Hepatic Bax inhibitor-1 inhibits IRE1alpha and protects from obesity-associated insulin resistance and glucose intolerance. J Biol Chem. 2010;285(9):6198-207
43. Jin R, Zhao A, Han S, Zhang D, Sun H, Li M. et al. The interaction of S100A16 and GRP78 actives endoplasmic reticulum stress-mediated through the IRE1α/XBP1 pathway in renal tubulointerstitial fibrosis. Cell Death Dis. 2021;12(10):942
44. White AM. Gender differences in the epidemiology of alcohol use and related harms in the United States. Alcohol Res. 2020;40(2):01
45. Sayaf K, Gabbia D, Russo FP, De Martin S. The role of sex in acute and chronic liver damage. Int J Mol Sci. 2022;23(18):10654
Author contact
![]() Corresponding authors: Yun Liu, Department of Geriatrics, The First Affiliated Hospital of Nanjing Medical University, 300 Guangzhou Road, Nanjing, 210029, China. Email address: liuyunedu.cn. Wen-Song Zhang, Department of Pharmacy, The First Affiliated Hospital of Nanjing Medical University, 300 Guangzhou Road, Nanjing, 210029, China. Email address: zhangwensong91com. Ke Jin, Department of Infectious Diseases, The First Affiliated Hospital of Nanjing Medical University, 300 Guangzhou Road, Nanjing, 210029, China. Email address: penghaoren2001com.
Corresponding authors: Yun Liu, Department of Geriatrics, The First Affiliated Hospital of Nanjing Medical University, 300 Guangzhou Road, Nanjing, 210029, China. Email address: liuyunedu.cn. Wen-Song Zhang, Department of Pharmacy, The First Affiliated Hospital of Nanjing Medical University, 300 Guangzhou Road, Nanjing, 210029, China. Email address: zhangwensong91com. Ke Jin, Department of Infectious Diseases, The First Affiliated Hospital of Nanjing Medical University, 300 Guangzhou Road, Nanjing, 210029, China. Email address: penghaoren2001com.