Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(7):1368-1382. doi:10.7150/ijbs.33233 This issue Cite
Research Paper
Exosomal Micro RNAs Derived from Dermal Papilla Cells Mediate Hair Follicle Stem Cell Proliferation and Differentiation
Hailong Yan1,2, Ye Gao1,3, Qiang Ding1, Jiao Liu1, Yan Li1, Miaohan Jin1, Han Xu1, Sen Ma1, Xiaolong Wang1, Wenxian Zeng1 ![]() , Yulin Chen1
, Yulin Chen1 ![]()
1. Key Laboratory of Animal Genetics, Breeding and Reproduction of Shaanxi Province, College of Animal Science and Technology, Northwest A&F University, Yangling, China
2. Life Science Research Center, Yulin University, Yulin, China
3. School of Medicine, Shanxi Datong University, Datong, China
Received 2019-1-17; Accepted 2019-3-24; Published 2019-5-20
Abstract

Recent studies have demonstrated that dermal papilla cell-derived exosomes (DPC-Exos) promote the anagen stage of hair follicle (HF) growth and delay the catagen stage. However, the roles of DPC-Exos in regulating hair follicle stem cell (HFSC) quiescence and activation remain unknown. Here, we found that HFSC differentiation was induced by co-culture with DPCs, and that DPC-Exos attached to the surface of HFSCs. Using micro RNA (miRNA) high-throughput sequencing, we identified 111 miRNAs that were significantly differentially expressed between DPC-Exos and DPCs, and the predicted target genes of the top 34 differentially expressed miRNAs indicated that DPC-Exos regulate HFSCs proliferation and differentiation via genes involved in cellular signal transduction, fatty acid expression regulation, and cellular communication. The overexpression of miR-22-5p indicated that it negatively regulates HFSC proliferation and LEF1 was revealed as the direct target gene of miR-22-5p. We therefore propose the miR-22-5p-LEF1 axis as a novel pathway regulating HFSC proliferation.
Keywords: exosome, hair follicle stem cells, dermal papilla, proliferation
Introduction
The Shaanbei White cashmere goat, a prominent Chinese domestic goat breed, is farmed to provide fine hair fiber (cashmere) and meat. Cashmere goats have primary hair follicles (HFs) and secondary HFs, which produce guard hairs and cashmere fibers, respectively [1]. Cashmere is an important economic trade good worldwide and has a global market [2]. The number and cyclical growth of HFs affect the diameter and yield of cashmere fibers [3], and incremental changes in HF stem cell (HFSC) behavior should result in effective changes in the size and structure of HFs. Due to their continuous cycles of quiescence and activation in different cell lineages, HFSCs represent an excellent model for studying the characteristics of adult somatic stem cell functions [4]. Understanding the proliferation and differentiation mechanisms of HFSCs is an important prerequisite for developing strategies designed to determine the principia of the plasticity and clinical therapy of hair loss diseases [5, 6].
Dermal papilla cells (DPCs) play an important role in the differentiation of HFSCs into HFs. In the resting phase, HFSCs located in the bulge region of the HFs. During the apoptosis of matrix cells, dermal papilla (DP) migrate upward and, when reaching the bulge, release signals that stimulate the differentiation of HFSCs and trigger the regeneration of HFs [7, 8]. Although Zhang et al. have previously constructed an in vitro co-culture system of DPCs and HFSCs using transwell inserts [9], the relationship between DPCs and HFSCs and the mechanisms whereby DPC-derived exosomes (DPC-Exos) regulate HFSC behavior are not fully understood.
Exosomes play an important role in cell-to-cell communication, and DPC-Exos have been shown to promote the anagen stage and delay the catagen stage of HF growth [10], and to stimulate the proliferation and differentiation of outer root sheath cells [11]. Exosomes transport micro RNA (miRNA) and mRNA or transcription factors (TFs) to nearby cells, thereby influencing the function of these cells [12-14]. Previous studies have indicated that exosomal miRNA plays an important role in regulating the target genes and signaling pathways that are necessary for osteoblast differentiation [15]. However, most of these studies have focused on cancer invasiveness and osteoblast differentiation, whereas the mechanisms whereby DPC-Exos miRNA regulates the proliferation and differentiation of HFSCs remain undetermined.
Micro RNAs are key negative regulating factors that influence the timing of organogenesis, apoptosis, proliferation, and differentiation [16, 17], and some play important roles in the regulation of HF development and periodic growth [18-22]. However, to date, no studies have examined the differences between the miRNA expression profiles of DPC-Exos and DPCs in HFs. Although exosomal miRNAs derived from DPCs might play a critical role in regulating the proliferation and differentiation HFSCs, this has yet to be determined. Thus, in the present study, we isolated and identified DPC-Exos from Shaanbei White cashmere goats, and sequenced DPC and DPC-Exos miRNAs via high-throughput sequencing to examine if these DPC-Exos miRNAs regulate the fate of HFSCs. To elucidate the regulatory mechanism of DPC-Exos miRNAs, we also identified the target genes of these specific miRNAs. The findings of this study will provide a theoretical reference for future research on the fine regulation of HFSC proliferation and differentiation.
Materials and Methods
Animals
Adult cashmere goats were obtained from the Shaanbei White Cashmere Goat Farm of Yulin University, Yulin, China. All studies were conducted in accordance with the guidelines approved by the Experimental Animal Committee of the Northwest A&F University, Yangling, China (Approval ID: 2014ZX08008-002).
Isolation and culture of HFSCs
The HFSCs isolated from the HFs of Shaanbei White cashmere goat adults were identified, preserved, and cultured as previously described [23]. Skin samples (1 cm2) were clipped from the back of adult Shaanbei White cashmere goats, sterilized in 3% povidone iodine and 75% ethanol, cut into 0.5 cm × 0.5 cm segments using ophthalmic scissors, washed at least three times with phosphate-buffered saline (PBS), and then treated with 0.25% Dispase II for 14 h at 4 ºC. The HFs were separated from the connective tissue using a syringe needle under a stereomicroscope, and cultured in 24-well dishes containing Collagen IV and Dulbecco's Modified Eagle Medium/ Nutrient Mixture F-12 (DMEM/F12) medium supplemented with 1% penicillin-streptomycin mixture, 10 ng/mL insulin, 10 ng/mL epidermal growth factor (EGF), 0.4 μg/mL hydrocortisone, and 2% fetal bovine serum (FBS) at 37ºC in a 5% CO2 atmosphere. After tissue adherence, the nutrient medium was changed every three days and cell growth was observed under the microscope.
Purification of HFSCs
Passage 1 and 2 (P1 and P2) HFSCs were digested with TrypLETM Express Enzyme (Gibco, Carlsbad, CA, USA) for 6-10 min at 37 °C, passaged every 6-10 days in a 1:3 to 1:5 ratio, and then purified using a modified version of a previously described method [24]. The purified P2 HFSCs were seeded in six-well plates and digested at 80-90% confluence. The reaction was terminated using fluorescence- activated cell sorting (FACS) buffer (500 mL Dulbecco's phosphate buffered saline + 3% FBS + 2 mL 0.5 mM ethylenediaminetetraacetic acid), and the cell suspension was then centrifuged at 1000 rpm for 5 min. After discarding the clear supernatant, the pelleted cells were resuspended in FACS buffer containing anti-CD34 primary antibody (1:100, ab81289; Abcam, Cambridge, UK), and incubated at room temperature for 40 min. Following incubation, the cells were washed twice in 3 mL FACS buffer and resuspended in FACS buffer containing Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody Alexa Fluor® 488 conjugate (1:1000, A-11008; Thermo Fisher Scientific, Waltham, MA, USA), and incubated on ice for 30 min. Cells were then washed twice with 3 mL FACS buffer and filter-centrifuged using 300-400 µm filters. The resulting pellet was resuspended in FACS buffer and placed on ice for FACS on the BD FACSAria™ III (BD Biosciences, San Jose, CA, USA), and the sorted cells were used for subsequent experiments. All cells between P4 and P6 were maintained at 37 °C under a 5% CO2 atmosphere for further experiments.
Cell growth curve
Fifth passage HFSCs were seeded at 1 × 104 cells/mL in 24-well plates and counted in triplicate at 24 h intervals using a hemocytometer. Mean cell numbers were calculated and used to plot cell growth curves.
Isolation culture and DiI labeling of DPCs
The DPCs were isolated as previously described [25-27]. Samples of Shaanbei White cashmere goat skin were collected and processed as described in “Isolation and culture of HFSCs”. After digestion for 30 min at room temperature in DMEM/F12 medium containing 0.2 mg/mL Collagenase II (Gibco), the DP of the HFs were microdissected using forceps and syringe needles under the microscope, and then added to a 24-well culture plate (one DP per well) and cultured in DMEM/F12 medium containing 10% FBS in 5% CO2 at 37 °C. After 3-5 days, the cells migrated from the mastoid and the medium was changed every 2 days. Only DPCs before P5 were used in subsequent experiments.
Following the addition of 1 μM DiI, the fluorescent dye used to label the DPCs, the cells were incubated at 37 °C for 1 h; this process was entirely performed in the absence of the light. Thereafter, the cells were washed five times with PBS and HFSCs were added to the culture medium. The recovered medium was used as the negative control in the subsequent experiment.
Co-cultivation of DPCs and HFSCs
We designed a pipeline to induce HFSCs differentiation and directly determine HF fate. The HFSCs were co-cultured with DPCs using transwell devices. In brief, after platting HFSCs in six-well plates, transwell inserts were added and then seeded with DPCs. All cells were cultured in fresh DMEM/F12 medium, and the HFSCs were harvested after 7 days. Total RNA and protein were extracted from at least three independent samples and analyzed using quantitative real-time PCR (qRT-PCR) and Western blotting, respectively.
To verify that the exosomes derived from the DPCs can be transported to HFSCs, after labeling the DPCs with DiI these were co-cultured with HFSCs in 0.4 μm transwells. As a negative control, HFSCs were incubated in the recovered medium used as the final wash for DiI-labeled cells, and observed under a fluorescence microscope after 24 h. After labeling exosomes with DiI, the incubated HFSCs were observed under a fluorescence microscope after 16 h.
Hematoxylin & eosin (H&E) staining
Some of the skin samples taken from the back of the Shaanbei White cashmere goat adults were biopsied in mid-March and mid-August. After harvesting, skin samples were fixed on a cardboard with pins on each corner. Tissue biopsies were fixed on the board in 4% paraformaldehyde, and then embedded in paraffin using routine histologic procedures. Four-micrometer sections were cut using a microtome, and, after H&E staining was carried out, longitudinal sections of HFs were obtained under a light microscope (40× and 100× amplification).
Transmission electron microscopy
The identified HFSCs were fixed with 2.5% glutaric PBS for 4 h or overnight. Thereafter, the cells were rinsed with PBS, fixed with 1% osmium tetroxide, rinsed with double-distilled water (ddH2O), fixed/stained with 2% uranyl acetate, and gradient dehydrated in 50%, 70%, 90%, 100% ethanol, and acetone, prior to infiltration, embedding, and polymerization. Sections of the embedded cells were cut using an ultramicrotome, followed by staining with uranyl acetate and lead citrate.
Isolated exosomes derived from DPCs were diluted 10 times, fixed in 4% paraformaldehyde, and then layered on a carbon/Formvar film-coated TEM grid (EMCN®, Beijing, China) for 20 min. The grids were treated with 1% glutaraldehyde and then washed eight times with ddH2O (2 min each time), and stained with 1% uranyl acetate for 10 min. The preparations were dried naturally and observed using a transmission electron microscope (Tecnai G2 Spirit Bio, FEI Company, Hillsboro, OR, USA). The Feret's diameter of the particles was determined using ImageJ software (Rasband, W.S., U.S. National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/, 1997-2014).
Quantitative real-time PCR
To identify the purified HFSCs, the mRNA expressions of K15, CD34, and ITGβ1 were determined by qRT-PCR. Total RNAs were extracted from samples using the RNeasy kit (Takara Biotechnology, Dalian, China) following the manufacturer's instructions. Complementary DNAs were then synthesized from 300 ng total RNA using the First Strand cDNA Synthesis kit (Thermo Fisher Scientific) according to the manufacturer's protocol. The qRT-PCR was performed in a CFX96 Detection System (Bio-Rad Laboratories, Hercules, CA, USA). Similarly, qRT-PCR was used to confirm the expression levels of S100A3 and K6 mRNA in HFSCs in which differentiation had been induced by DPCs.
To validate the miRNA expression data obtained from sequencing, the expression of eight miRNAs (miR-1, miR-122, miR-378-3p, miR-145-5p, let-7f-5p, miR-21-5p, miR-592, and miR-106a-5p) was determined in six samples (three DPC and three DPC-Exos samples) using qRT-PCR. Small RNAs were extracted from cultured cells and exosomes as described above and then spiked with synthetic cel-miR-39 as an internal loading control for analysis of miRNA expression using qRT-PCR [28]. The primer sequences used for qRT-PCRs are listed in Supplementary Table S1. All qRT-PCR reactions were repeated three times, independently.
miRNA library construction and sequencing
Small RNAs from DPCs and DPC-Exos were used for miRNA library construction and sequencing, which were performed by Novogene Co., Ltd., Beijing, China, using 20 ng small RNA per sample as the input material. Sequencing libraries were constructed following the recommendations of the manufacturer of NEBNext® Multiplex Small RNA Library Prep Set (NEB, Ipswich, MA, USA) for Illumina® (San Diego, CA, USA). Small RNAs were reverse transcribed and then amplified in the Illumina HiSeq 2500 platform that generated 50-bp single-end reads for sequencing. Sequencing data were trimmed using Cutadapt to remove the reads containing poly-N, 5′ adapter contaminants, poly A, T, G, or C, reads without a 3′ adapter or insert tag, and low quality reads [30], and then aligned against the goat reference database (GCF_001704415.1_ARS1_genomic.fna) using bwa-mem [31]. Known miRNAs were detected by using Mapped small RNA tags and the miRBase21 as reference. To remove tags originating from protein-coding genes, repeat sequences, ribosomal RNA (rRNA), transfer RNA (tRNA), small nuclear RNA (snRNA), and small nucleolar RNA (snoRNA), small RNA tags were mapped to RepeatMasker and Rfam databases or to these types of data from cow and sheep species. In alignment and annotation, some small RNA tags might be mapped to more than one category. Accordingly, to ensure that each small RNA was mapped to only one annotation, we adopted the following priority rule: miRNA > rRNA > tRNA > snRNA > snoRNA > novel miRNA.
Novel miRNAs were predicted using mireap via analyses of the secondary structure, Dicer cleavage site, and minimum free energy of the small RNA tags unannotated in the previous. Micro RNA expression levels were estimated in terms of transcripts per million (TPM) based on the following criterion [32]: Normalization formula, Normalized expression = mapped read counts /Total reads × 1,000,000. Differential expression analysis of two conditions/ groups was performed using the DESeq R package (1.20.0), adjusting the P-value with the q value [33]. A q value <0.05 and |log2 (fold change)| >1 were set as the default thresholds for significantly different expression.
Target gene prediction, and gene ontology and Kyoto Encyclopedia of Genes and Genomes enrichment analyses
Prediction of the target genes of the top 34 miRNAs was performed using RNA22 (R22) (Version 2.0) [34] and Pita (Version 6.0) [35]. Target gene candidates of differentially expressed miRNAs were screened for gene ontology (GO) enrichment (hereafter referred to “target gene candidates”). GOseq-based Wallenius non-central hyper-geometric distribution [36], which can compensate for gene length bias, was implemented for GO enrichment analysis. We used KOBAS [37] software to determine the enrichment of the target gene candidates in Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways.
Immunofluorescent staining
To analyze HFSCs by immunofluorescent staining, the cells were first fixed and blocked with 4% paraformaldehyde, and then incubated with the following primary antibodies: anti-K15 (1:100, ab111448; Abcam), anti-ITGβ1 (1:100, ab155145; Abcam), and anti-CD34 (1:100, ab81289; Abcam). After this incubation, HFSCs were incubated with fluorescein isothiocyanate-conjugated secondary antibodies and counterstained with 4′,6-diamidino-2- phenylindole (DAPI; Sigma-Aldrich, St. Louis, MI, USA). Culture plates were visualized using a fluorescence microscope (AMG, Mill Creek, WA, USA).
Western blot analysis
The HFSCs and DPCs were lysed in 250 μL radioimmunoprecipitation assay (RIPA) buffer containing 1 mM phenylmethane sulfonyl fluoride (PMSF) (both Beyotime Biotech, Jiangsu, China). The lysed cells were centrifuged, and the resulting supernatants mixed with 4× sodium dodecyl sulfate (SDS) buffer containing 50 mM dithiothreitol. After boiling, the samples, together with protein standards (#1610374; Bio-Rad), were subject to 12% SDS- polyacrylamide gel electrophoresis (PAGE) in running buffer at 90 V for 2 h. Following separation, the proteins were electrotransferred onto polyvinylidene difluoride membranes, which were then incubated with TBST (50 mM Tris, 150 mM NaCl, 0.05% Tween-20) for 1 h at room temperature. The membranes were probed with anti-K15 (1:1000, ab111448; Abcam), anti-ITGβ1 (1:1000, ab155145; Abcam), and anti-CD34 (1:1000, ab81289; Abcam), and their binding was detected using an enhanced chemiluminescence reagent (GE Healthcare Bioscience, Chicago, IL, USA) following the manufacturer's instructions. To ensure that similar levels of total proteins were loaded onto gels, the membranes were stained with Swift Membrane Stain (G Biosciences, St. Louis, MO, USA) according to the manufacturer's instructions. An imaging system (ChampChemi™ Top 610, Sage Creation Science, Beijing, China) was used to visualize the protein bands.
Exosome samples were also analyzed following lysis in SDS buffer containing 50 mM dithiothreitol with boiling for 5 min at 100 ºC. For detection, membranes were probed with anti-CD63 (25682-1- AP) and anti-Canx (10427-2-AP) (both Proteintech Group, Inc., Rosemont, IL, USA).
Exosome isolation and labeling
Exosomes were isolated from 40 mL serum-free culture medium containing DPCs via a series of differential ultracentrifugation steps as described previously [29]. The DPCs were cultured in 10% FBS medium until cell density reached 60%, washed five times with DPBS (Hyclone), and then cultured with 10% knockout serum replacement (KSR) (A3181502, Thermo Fisher Scientific) medium. After 48 h, the medium was collected and gradient-centrifuged; first at 300 ×g for 10 min to eliminate residual cells, and then at 2000 ×g for 10 min to eliminate dead cells. To remove cell debris, the medium was finally centrifuged at 10,000 ×g for 30 min. Thereafter, the supernatant was collected and filtered through a 0.22-μm membrane to remove large particles. Ultracentrifugation was performed at 100,000 ×g for 70 min to pellet the DPC-Exos. The pellets were then washed in PBS at 100,000 ×g for 70 min. All centrifugations were performed at 4 ºC. The resulting pellets were resuspended in PBS for further analysis. The isolated DPC-Exos were labeled for 1 h with 1 μM DiI in a 37 ºC water bath; the entire process was conducted in the dark. Following the addition of PBS to fill the centrifuge tubes, the labeled exosomes were centrifuged at 100,000 ×g for 70 min at 4 ºC. After resuspended the resulting pellet in 100 μL PBS, the tubes were wrapped in tin foil and stored at -80 ºC.
Nanoparticle tracking analysis
To determine the particle size and concentration of DPCs within 10% KSR medium, we performed nanoparticle tracking analysis (NTA) using a NanoSight LM10 instrument (Malvern Instruments, Malvern, UK). Initially, exosome preparations were diluted in PBS to obtain an ideal concentration of 106 to 108 particles/mL. Each analysis required at least 300 μL of diluted sample, and this was mixed by vortexing before being injected into the chamber. Three videos of diluted samples were recorded and the resulting counts were averaged for each diluted sample (three replicates per sample). The duration of moving particles was 60 s for each video, with a shutter speed of 30 ms and camera gain of 680. The data were analyzed using NTA 2.3 analytical software (Malvern Instruments).
Cell proliferation assays
To confirm the effects of miR-22-5p on HFSCs, either miR-22-5p mimics or negative control (NC) (GenePharma, Shanghai, China) were transfected into HFSCs using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions. Each sample was transfected in triplicate. Cell proliferation was measured using the CCK-8 kit (Dojindo Molecular Technologies, Inc., Kumamoto, Japan), the EdU Cell Proliferation kit with Alexa Fluor 488 Imaging kit (Thermo Fisher Scientific), and cell cycle. The cells were seeded into 96-, 48-, and six-well microplates at a density of 5 × 103, 1 × 104, and 5 × 104 cells per well. Forty-eight hours after transfection with miRNA-22-5p mimics or NC, cell proliferation was detected using the CCK-8 kit, EdU kit, or cell cycle following the manufacturer's instructions. The absorbance of samples was measured at 450 nm using a plate reader.
Statistical analyses
All results are expressed as the mean ± standard deviation (SD) of at least three independent experiments. Microsoft Excel was used for statistical analyses. The statistical significance of differences was evaluated using paired t-tests and considering P <0.05 as statistically significant.
Results
Isolation and identification of HFSCs from Shaanbei White cashmere goats
Within the HFs, HFSCs are localized in a specialized niche, referred to as the bulge, and contribute to HF regeneration and wound repair [38]. They can also differentiate to epidermal and sebaceous gland cells, and eight different types of HF epithelial cells [39]. To obtain the desired HFSCs, single cashmere goat HFs were isolated and used for adherent culture. After 3 days, few cells had migrated from the bulge region but this number increased by the sixth day of culture (Figure 1A). The CD34+ cells were isolated and purified by flow cytometry to obtain CD34+ cells with purity over 90% (Figure 1B). Morphologically, these cells were small and round with a three-dimensional nest-like shape, refractive, and were characterized by a cobblestone-like pavement growth. Within each cell, a centralized nucleus containing more than two nucleoli was observed, as well as a high nuclear-cytoplasmic ratio. These are typical morphological characteristics of skin stem cells (Figure 1C, 1D). The isolated and purified cells proliferated slowly during the first 1 to 3 days of incubation, but thereafter began to proliferate rapidly and entered the logarithmic growth phase. On the eight day, the proliferation gradually slowed down and cell growth entered the plateau phase (Figure 1E).
Isolation and characterization of hair follicle stem cells (HFSCs) from the Shaanbei White cashmere goat. A. Adhesion culture of hair follicles. Scale bar, 1000 μm. B. Fluorescence-activated cell sorting of CD34. C. HFSCs after purification. Scale bar, 1000 μm. D. Observation of HFSCs by transmission electron microscopy. Scale bar, 2 μm. E. Growth curve of HFSCs. Passage five HFSCs were seeded in 24-well plates at a density of 1 × 104 cells/mL and cell counts were performed at 24 h intervals using a hemocytometer. Three wells were counted for each time point, and the mean cell number was calculated to plot a cell growth curve. F. Protein expression of HFSC markers. G. Immunocytochemical staining of HFSCs markers. Scale bar, 100 μm. H. Relative mRNA expression levels of HFSC markers.
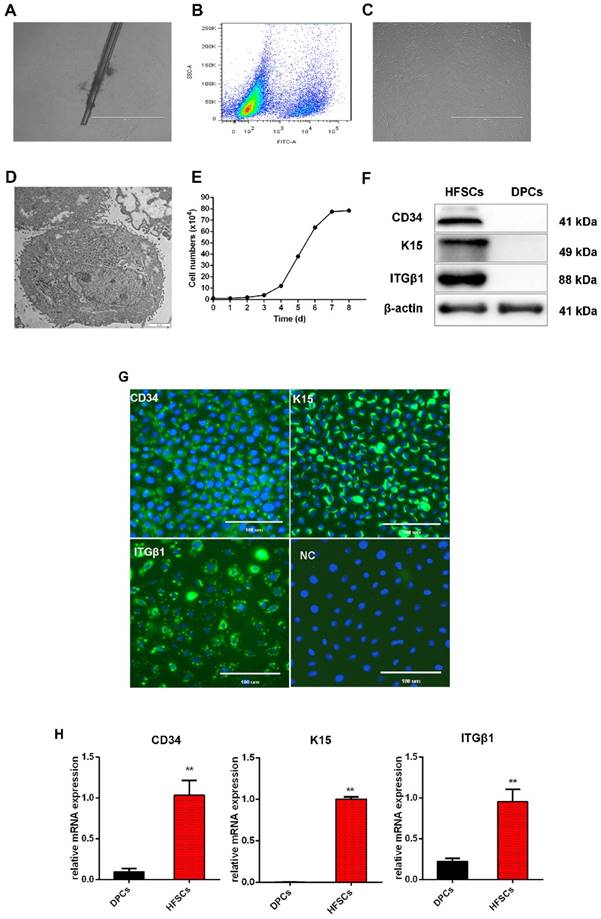
To characterize these cashmere goat bulge- derived cells, the expressions of the HFSC markers CD34, K15, and ITGβ1 were analyzed at both the mRNA and protein levels. Positive expression of CD34, K15, and ITGβ1 was detected by Western blotting (Figure 1F) and immunofluorescence (Figure 1G) analyses, and we found that the mRNA expression levels of CD34, K15, and ITGβ1 in HFSCs were significantly increased compared with those in DPCs (P <0.05) (Figure 1H). To further verify the differentiation capacity of the bulge-derived cells, these were induced to differentiate via co-culture with DPCs. The marker mRNAs of HFSCs cultured in normal medium and co-cultured with DPCs were detected by qRT-PCR. The results showed that, compared with HFSCs cultured in isolation, the expression of the HF-specific markers S100A3 and K6 in the HFSCs co-cultured with DPCs were significantly increased (P <0.05) (results not shown). These data indicated that the cells isolated from HFs exhibited HFSC properties and differentiation potential. Therefore, the in vitro culture system established for HFSCs in this study can be used in further research.
Identification and transport of exosomes in DPCs
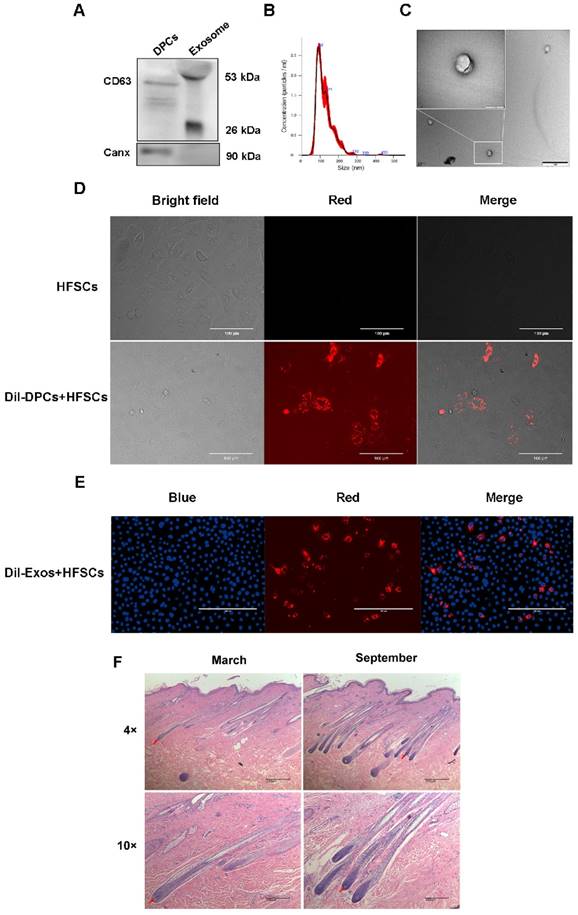
By inducing the differentiation of HFSCs, DPCs play a key role in regulating hair growth [9, 40, 41], and DPC-Exos may be important regulatory factors in this process. Therefore, DPC-Exos were isolated and identified in the present study. To examine the purity of the DPC-derived extracellular vesicles (DPC-EVs), we employed several different methods. The results of Western blot analysis showed that the exosome-specific marker CD63 was detected in DPCs-EVs, whereas the cell marker Canx was not (Figure 2A). The NTA revealed that the average size of DPC-EVs was 123.8 ± 0.6 nm (Figure 2B). Transmission electron microscopy revealed that DPC-EVs were ~100 nm and the presence of round EVs, as well as EV membranes (Figure 2C), whereas no similar structure was identified in DPBS or in the culture medium containing serum substitutes (results not shown). Accordingly, we were able to verify that these EVs were exosomes. To analyze the uptake of DPC-Exos into HFSCs, we labeled the DPCs and DPC-Exos with the fluorescent dye DiI and then co-cultured the DiI-labeled DPCs with HFSCs for 24 h using a transwell system; the DiI signals on HFSCs were observed using fluorescence microscopy. The HFSCs were also incubated in the medium that had been used as a final wash in the preparation of DiI-labeled DPCs, but no fluorescence signals were detected excluding the possibility of non-specific staining (Figure 2D). Fluorescence microscopy revealed the uptake DiI-labeled DPC-Exos by HFSCs (Figure 2E), and that the exosomes present in DPCs could be internalized by HFSCs. To examine cell morphological differences during the HF cell cycle, we collected skin samples in the anagen (mid-September) and the telogen (mid-March) phases, which were determined according to HF morphological characteristics. The H&E staining showed that the size of DPs in secondary HFs changed dramatically between anagen and telogen phases, and that HF undergo a structural change that brings DP closer to HFSCs in the telogen phase (Figure 2F). Current evidence suggests that the proximity between DP and HFSCs is necessary for HFSCs activation and generation of a new hair [42-44]. Taken together, these results indicated that DPCs migrate close to the bulge where HFSCs are located during the telogen phase and may regulate HFSCs by deriving exosomes to generate new hair germ cells.
Identification and functional analysis of exosomes derived from dermal papilla cells (DPCs). A. Detection of CD63 and Canx in exosomes. B. Analysis of exosome size by nanoparticle tracking analysis. C. Observation of DPC-derived exosomes by transmission electron microscopy. Scale bar, 1 μm. D. Transport of extracellular vesicles (EVs) in co-culture without physical contact between different cell types. Transwell systems were used to separate recipient cells from DiI-labeled DPCs, with DiI-labeled DPCs in the upper filter and hair follicle stem cells (HFSCs) in the lower chamber. Scale bar, 100 μm. E. DiI-labeled exosomes derived from DPCs (DPC-Exos) were taken up by HFSCs (DiI label is shown in red, 4′,6-diamidino-2-phenylindole label is shown in blue). Scale bar, 200 μm. F. Hematoxylin and eosin staining results of goat hair cells in the anagen and telogen phases. Red arrows show the location of dermal papillae. Scale bar, 200 μm.
miRNA expression profile of DPC-Exos
Exosomes are known to contain miRNAs, which can be delivered to other cells thereby affecting cellular function [45]. Because miRNA expression profiles vary according to cell type and growth conditions [46], in the present study, we sought to investigate the miRNA expression profiles of exosomes derived from DPCs using Illumina HiSeq high-throughput sequencing. Among the 77,144,497 raw reads produced, 69,930,479 were clean reads, and the clean read generation rate was higher than 91% for all samples.
To identify known miRNAs, the clean reads from DPCs and DPC-Exos libraries were compared with the goat genome reference sequence. The remaining reads were used to predict novel miRNAs (Supplementary Table S2). To focus on the highly represented miRNAs, unique reads were normalized and miRNAs with a TPM value lower than 0.5 in three samples were omitted from the analysis. Finally, 271 types of known miRNAs and 336 predicted novel miRNAs were identified (Supplementary Table S3, Supplementary Table S4). We found high similarity in the expression patterns of both DPC and DPC-Exos samples (Figure 3A). The number of overlapping and unique miRNAs between DPCs and DPC-Exos are shown in Figure 3B. The raw sequencing data generated here were submitted to the National Center for Biotechnology Information (NCBI) sequence read archive (SRA) under accession number PRJNA511763.
miRNA expression profiles of exosomes derived from dermal papilla cells (DPC-Exos). A. Schematic diagram of relative miRNA expression between samples. X- and Y-axes represent the sample log10 (TPM+1) and the square of Pearson correlation coefficients (R2), respectively. B. Venn diagram showing the unique and overlapping miRNAs between DPCs and DPC-Exos. Numbers in parentheses are the numbers of differentially expressed miRNAs in DPCs and DPC-Exos. C. Heatmap diagram of differential miRNA expression between DPCs and DPCs-Exos. Red, blue, and white colors indicate increased expression, decreased expression, and mean values, respectively. Each row in the figure represents one miRNA and each column shows one sample. Each cell shows the differential expression of each miRNA. D. Volcano plot of known differential miRNAs. The X-axis represents the miRNA fold change in different groups or samples, whereas the Y-axis represents the statistical significance of the change in miRNA expression. Circles represent miRNA: green circles denote miRNAs that were not significantly expressed, red circles denote significantly up-regulated miRNAs, blue circles denote significantly down-regulated miRNAs. E. Volcano plot of novel differential miRNAs. F. Quantitative real-time PCR validation of miRNAs expression between DPCs and DPC-Exos. The abundances of miR-1, miR-122, miR-378-3p, miR-145-5p, let-7f-5p, miR-21-5p, miR-592, and miR-106a-5 were normalized to the abundance of miRNA 39 (* P <0.05).
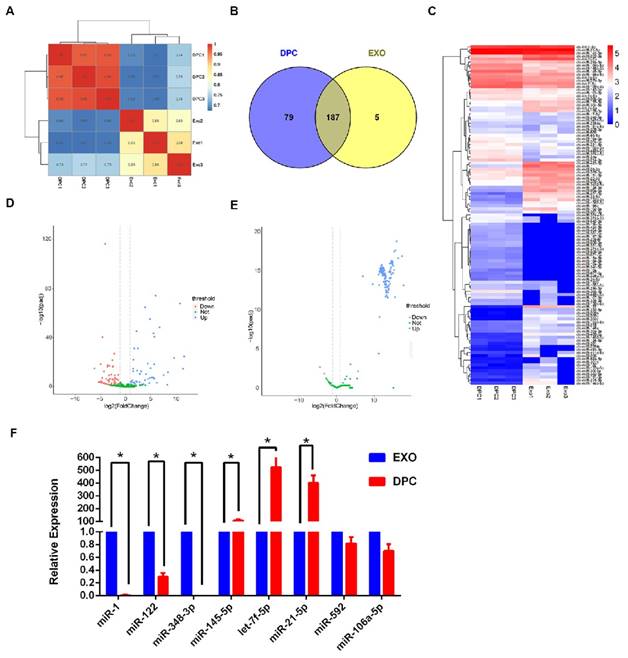
When we compared the expression levels of miRNAs in DPC-Exos and DPCs, using a |log2 (fold change)| >1 and adjusted P <0.05 as the threshold cutoffs, we found that among the 271 known miRNAs, 111 showed significantly different expression between DPC-Exos and DPCs (Figure 3C, Supplementary Table S5). We detected the expression of 142 novel miRNAs in DPC-Exos and DPCs, which are depicted in the volcano plots shown in Figure 3D and 3E. Collectively, these data indicated that exosomal miRNAs were differentially expressed in DPC-Exos and DPCs.
To verify the sequencing data, we selected three miRNAs (miR-1, miR-122, and miR-348-3p) that were up-regulated in DPC-Exos, three miRNAs (miR-145- 5p, let-7f-5p, and miR-21-5p) that were down- regulated in DPC-Exos, and two undifferentiated miRNAs (miR-592 and miR-106a-5p) from the miRNA array. Consistent with the miRNA sequencing data, the qRT-PCR showed similar trends of miRNA expressions (Figure 3F). These data indicated that our preliminary screening assay for differentially expressed miRNAs in exosomes was reliable and feasible. Further, these miRNAs may play a functional role in exosomal miRNA signatures related to HFSC biological processes.
DPC-Exos miRNAs with altered gene expression are involved in cellular processes and hair morphogenesis pathways
To determine which cellular and biological pathways are potentially regulated by exosomal miRNAs derived from DPCs, we performed a comprehensive comparison between the predicted target genes for the top 34 differentially expressed miRNAs, 22 and 12 of which were significantly up- and down-regulated in DPC-Exos, respectively (Supplementary Table S6).
To gain a better understanding of the gene networks in DPC-Exos, we performed GO and KEGG analyses. The differentially expressed genes were found to be significantly enriched in 50 GO terms associated with molecular function, 60 GO terms associated with biological processes, and nine GO terms associated with cell components. Additional analysis revealed that 93.9% of the genes were involved in molecular function and 53.1% of the genes were related to binding (Figure 4A, Supplementary Table S7). Differentially expressed genes were enriched in KEGG pathways related to 40 biological functions, and mostly to HF intercellular communication including axon guidance, glycosylphosphatidylinositol (GPI)-anchor biosynthesis, endocytosis, fatty acid metabolism, and Ras signaling pathway (Figure 4B, Supplementary Table S8). These findings indicated that the identified pathways were involved in the proliferation and differentiation of HFSCs and hair development.
Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses. A. Histogram depicting candidate target gene GO enrichment. The X-axis represents the number of target genes associated with a unique term (and corresponding sub-terms). The Y-axis denotes the GO terms. The three different classifications represent the three basic GO term classes (from top to bottom, biological process, cellular component, and molecular function). B. Candidate target gene KEGG enrichment scatter plot. The X-axis represents pathways and the Y-axis represents the enrichment factor. The size of dots indicates the number of target genes in a pathway, and the color of dots indicates the different range of q-values. C. Enrichment map of biological processes targeted by the top 34 differentially expressed miRNAs in DPC-Exos. Nodes represent the 29 genes associated with HFSCs and hair shaft growth and the nine candidate miRNAs that target them, which are connected by the edges.
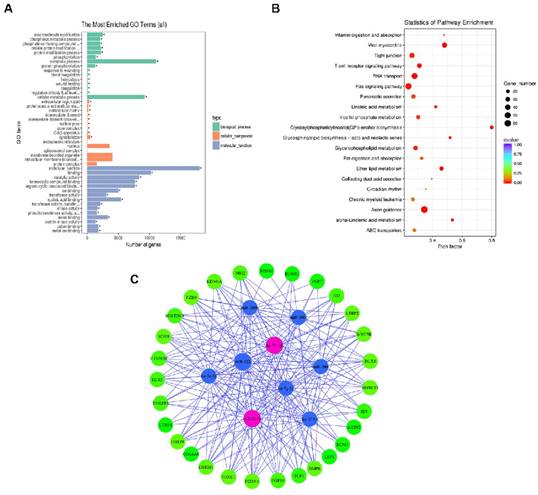
To gain further insights into the role of DPC-Exos in HF development related to the miRNA/target gene interactive networks, we determined the interaction network of nine candidate miRNAs among the selected 34 miRNAs and 29 differentially expressed genes associated with HFSCs and hair growth [47-53] (Figure 4C, Supplementary Table S9). Among the top 25 upregulated miRNAs in our screening, miR-let-7b-5p and miR-22-5p were identified as exosomal-enriched miRNAs that target genes related HF fate, including Dlx3, Foxn1, and Hoxc13, and the Wnt/Bmp antagonist Sostdc, respectively [54].
Exosomal miR-22-5p derived from DPCs inhibited HFSC proliferation and targeted LEF1
The expression of miR-22-5p was significantly higher in DPC-Exos than in DPCs and targeted multiple genes critical for HF and hair shaft formation [54]. To determine whether exosomal miR-22-5p influences HFSC proliferation, we performed CCK-8 and EdU assays, and showed that the proliferation of HFSCs transfected with miR-22-5p mimics was significantly decreased (P <0.01) compared with those transfected with NC (Figure 5A-5C). Subsequently, cell cycle phase distribution was analyzed using flow cytometry, and the results also revealed that overexpression of miR-22-5p induced HFSC cycle arrest at the S phase (Figure 5D-5E). Collectively, these results indicated that miR-22-5p inhibits HFSC proliferation.
miR-22-5p inhibits hair follicle stem cell (HFSC) proliferation by targeting LEF1. A. CCK-8 was used to detect the effect of miR-22-5p on HFSCs. B-C. The statistical results and representative figures of the effect of miR-22-5p on the proliferation of HFSCs as detected by EdU. ** P <0.01; Scale bar, 400 μm. D-E. Effect of miR-22-5p on the cell cycle of HFSCs. * P <0.05. F. Dual-luciferase reporter assay. * P <0.05, ** P <0.01.
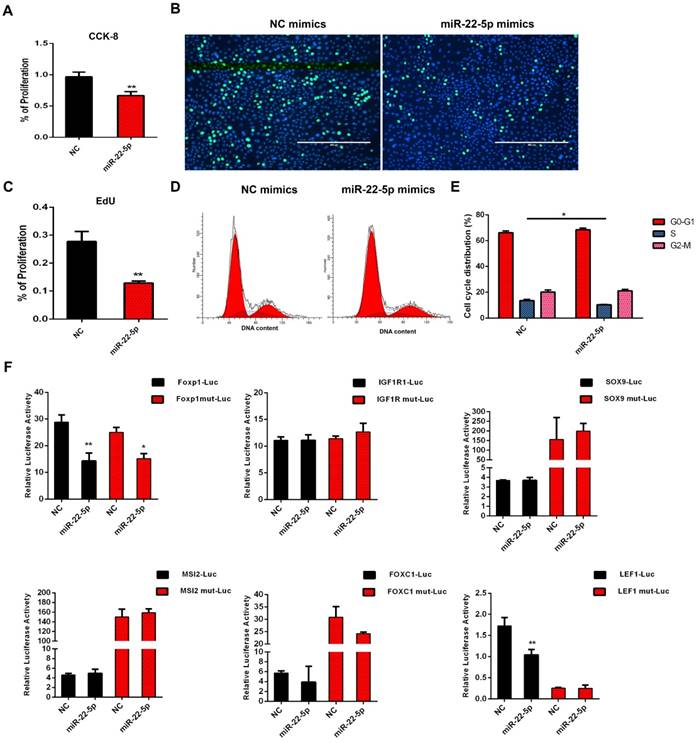
To further examine the molecular mechanisms underlying the inhibition of HFSC proliferation by miR-22-5p, we screened genes LEF1, FOXP1, IGF1R, SOX9, MSI2, and FOXC1 based on the results of miR-22-5p target gene prediction, and analyzed their activities using a dual luciferase reporter assay. The luciferase activities of FOXP1 and LEF1 were significantly decreased after transfection with miR-22-5p mimics. However, mutation of the target gene binding-site was found to result in recovery of the luciferase activity of LEF1, whereas the luciferase activity of FOXP1 was still significantly inhibited. Furthermore, we detected no interaction between miR-22-5p and the predicted target sites of IGF1R, SOX9, MSI2, and FOXC1, thus suggesting that LEF1 is the target gene of miR-22-5p, and that miR-22-5p can regulate LEF1 via the predicted target site (Figure 5F). Taken together, these results indicated that DPC-Exo miR-22-5p inhibits HFSC proliferation and targets the LEF1 gene.
Discussion
Cashmere fiber growth is a seasonal phenomenon characterized by a number of changes in HF morphology [55]. Hair follicle morphogenesis and cycling are regulated by complex and intricate interactions between DPCs and HFSCs, and require the spatiotemporal integration of multiple stimulatory and inhibitory signals [56-59], among which the WNT and BMP/TGFβ pathways play important roles [59, 60]. The DPC regulation of HF growth is mediated via a paracrine mechanism, in which exosomes may play a seminal role [11, 61]. Although DPC-Exos are known to promote the anagen stage of HF growth and delay the catagen stage [10], the role of DPC-Exos in regulating HFSC fate has yet to be determined. In the present study, we successfully isolated cashmere goat DPC-Exos from DPCs culture media that were consistent in terms of size and shape with exosomes reported in previous studies [62] and demonstrate that DPC-Exos can be transported into HFSCs.
With the continuing development of bioinformatics, the selection of candidate genes and miRNAs associated with periodic growth is becoming a common trend in cashmere goats [18-20]. However, previous high-throughput studies have tended to examine only HF or single-type cells. Although recent evidence indicated that exosomes are implicated in HF periodic growth [63], there is a lack of information concerning the regulatory role played by DPC-Exos miRNAs. To the best of our knowledge, the present study is the first in which the miRNA expression profiles of DPC-Exos have been verified using miRNA high-throughput sequencing. We identified five miRNAs (miR-200b, miR-200c, miR-330-5p, miR-200a, and miR-133a-5p) that are expressed in DPCs-Exos but not in DPCs. Among these, previous research has indicated that miR-200b and miR-200c are highly expressed in skin epidermal cells and that this expression is related to epithelial-mesenchymal transition [21, 22], highlighting the emerging role of the miR-200 family of miRNAs in cancer metastasis and epithelial-mesenchymal transition. Furthermore, some miRNAs may be particularly packed into exosomes and it has been suggested that the profile of miRNAs derived from exosomes does not reflect that of the donor cell [64]. In the present study, we determined exosomal miRNA profiles for comparison with cellular miRNA expression profiles. These unique exosomal miRNAs may play essential roles in the complex and intricate interactions between DPCs and HFSCs.
Exosomal miRNA-targeted genes provide an experimental basis for further research on the regulatory mechanisms of miRNAs [65]. Accumulating data indicate that certain miRNAs, including miR-214 and miR-205, are implicated in HFSC proliferation and differentiation by targeting the Wnt/β-Catenin and PI(3)K signaling pathways in vitro [66, 67]. To date, however, the function of the exosomal miRNA-mRNA network at the molecular level has not been fully determined. Twenty-nine of the target genes identified here are associated with HFSCs and hair shaft growth [47-53]. In the present study, we identified axon guidance signaling as the most significant pathway, as it was associated with 62 of the identified target genes. Axon guidance signaling was enriched in multiple signatures, implying that these factors play important roles in driving the large-scale cellular rearrangements necessary for HF formation and orientation [68, 69]. Recent studies have reported that fatty acids can function as ligands for peroxisome proliferator- activated receptors, promoting keratinocyte differentiation [70]. In this regard, in addition to containing unique miRNAs, exosomes are also characterized by a specific composition of lipids and proteins [71], and in the present study, we found that the miRNAs expressed in DPC-Exos are related to lipid and fatty acid metabolism signaling pathways.
Exosomes, as important components of paracrine signaling, can mediate the communication between distant cells via the direct transfer of various biomolecules, including miRNAs, from donor to recipient cells [72]. In the present study, we identified some of the mechanistic roles of these organelles in cellular communication, including their involvement in lipid rafts, the Ras signaling pathway, and endocytosis. We found that 80% of the genes associated with the GPI-anchor biosynthesis signaling pathway are target genes of exosomal miRNAs, and we identified lipid rafts as playing potential roles in HF epithelial-mesenchymal interactions [73]. The Ras signaling pathway is enriched in exosomes [74], thereby indicating the mechanistic role of exosomes in cellular activation and that exosomal miRNAs might exert a regulatory function in determining HFSC fate through networking with these cellular communication signaling pathways. Many paracrine factors and signaling pathways involved in this crosstalk at different stages of the hair cycle have been reported [75, 76], and the findings of the present study will facilitate further analysis to determine how DPC-Exos regulate cellular interactions through paracrine mechanisms.
Recent research has indicated that miR-22 represses hair neogenesis and HFSC colony formation capacity, and directly inhibits numerous transcription factors upstream the phenotypic keratin genes, including Hoxc13, Dlx3, and Foxn1 [54]. Notably, we found that DPC-Exos miRNA-22 consists of two forms, miR-22-5p and miR-22-3p; however, whereas the log2 fold change of miR-22-5p was 4.4, that of miR-22-5p was 1.74, indicating that these two forms of miRNA-22 may play different roles in HFSC development. It has been demonstrated that DPC-Exos stimulate the expression of β-catenin, which regulates HF development [11], whereas the activation of Wnt signaling modulates inductive signals in HF epithelial-mesenchymal interactions, thereby positively regulating HF growth and inducting and maintaining the anagen phase and hair cycle regeneration [77, 78]. Our observations suggest that DPC-derived exosomal miR-22-5p inhibits Wnt signaling expression. Although the precise mechanisms underlying the action of DPC-Exos in HF development remain to be clarified, we verified that the miR-22-5p produced by DPCs-Exos can regulate HFSC proliferation through targeting LEF1, a key transcriptional factor of the Wnt signaling pathway that directs HF patterning and stem cell fate [79, 80]. Zhang et al. demonstrated that LEF1 plays a key role in HFSC differentiation, promoting β-catenin translocation, thereby determining HFSC-directed hair fate [9]. Accordingly, in the present study, we demonstrated that the miR-22-5p-LEF1 axis may constitute a novel pathway for regulating HFSC proliferation. Our findings provide a platform for further enhancing the intrinsic regeneration of HFSCs, which would provide a valuable basis for the development of clinically viable exosome-based therapeutics.
In summary, exosomal miRNAs play a key role in regulating the target genes and signaling pathways that are essential for HFSC proliferation and differentiation, which in turn affect the yield and diameter of cashmere fibers. In the present study, we determined the miRNA expression profiles of exosomes derived from the DPCs of cashmere goat skin, and identified 111 miRNAs that were differentially expressed in the DPC-Exos compared with DPCs. We further revealed that miR-22-5p suppresses HFSC proliferation and targets LEF1 thereby establishing a new layer in the regulation of HFSC proliferation and differentiation by DPC-Exos miRNAs. Our findings also provide new insights on how to modify the direction HFSC differentiation for tissue engineering purposes.
Supplementary Material
Supplementary table s1.
Supplementary table s2.
Supplementary table s3.
Supplementary table s4.
Supplementary table s5.
Supplementary table s6.
Supplementary table s7.
Supplementary table s8.
Supplementary table s9.
Abbreviations
HF: hair follicles; HFSCs: hair follicles stem cells; DP: dermal papilla; DPCs: dermal papilla cells; DPC- Exos: dermal papilla cell derived exosomes; miRNA: micro RNA; P: passage; FACS: fluorescence-activated cell sorting; qRT-PCR: quantitative real-time PCR; H&E: Hematoxylin & eosin; RIPA: radioimmunoprecipitation assay; PMSF: phenylmethane sulfonyl fluoride; PAGE: polyacrylamide gel electrophoresis; rRNA: ribosomal RNA; tRNA: transfer RNA; snRNA: small nuclear RNA; snoRNA: small nucleolar RNA; NTA: nanoparticle tracking analysis; TEM: transmission electron microscopy; TPM: terms of transcripts per million; GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; EVs: extracellular vesicles; TFs: transcription factors.
Acknowledgements
We are grateful to Lei Qu and Jinwang Liu of Yulin University and Nimantana He of Inner Mongolia Agricultural University for the kind help and cooperation during the animal experiments. This work was supported by grants from the National Natural Science Foundation of China (31772571, 31572369, 31872332, 31760650), Key Research Program of Shaanxi Province (2017NY-072), as well as by China Agriculture Research System (CARS-39).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ansari-Renani HR, Ebadi Z, Moradi S, Baghershah HR, Ansari-Renani MY, Ameli SH. Determination of hair follicle characteristics, density and activity of Iranian cashmere goat breeds. Small Ruminant Research. 2011;95:128-32
2. Haiying L, Chao L, Guiqin Y, Hui L, Jin D, Yuyan C. et al. DNA Polymorphism of Insulin-like Growth Factor-binding Protein-3 Gene and Its Association with Cashmere Traits in Cashmere Goats. Asian-Australasian Journal of Animal Sciences. 2012;25:1515
3. McDonald BJ, Hoey WA, Hopkins PS. Cyclical fleece growth in cashmere goats. Australian Journal of Agricultural Research. 1987;38:597-609
4. Blanpain C, Fuchs E. Plasticity of epithelial stem cells in tissue regeneration. Science. 2014;344:1242281
5. Harries MJ, Meyer K, Chaudhry I, E Kloepper J, Poblet E, Griffiths CE. et al. Lichen planopilaris is characterized by immune privilege collapse of the hair follicle's epithelial stem cell niche. The Journal of pathology. 2013;231:236-47
6. Gilhar A, Etzioni A, Paus R. Alopecia areata. New England Journal of Medicine. 2012;366:1515-25
7. Zhang B, Tsai P-C, Gonzalez-Celeiro M, Chung O, Boumard B, Perdigoto CN. et al. Hair follicles' transit-amplifying cells govern concurrent dermal adipocyte production through Sonic Hedgehog. Genes & Development. 2016;30:2325-38
8. Reynolds AJ, Oliver R, Jahoda C. Dermal cell populations show variable competence in epidermal cell support: stimulatory effects of hair papilla cells. Journal of cell science. 1991;98:75-83
9. Zhang Y, Yu J, Shi C, Huang Y, Wang Y, Yang T. et al. Lef1 contributes to the differentiation of bulge stem cells by nuclear translocation and cross-talk with the Notch signaling pathway. International journal of medical sciences. 2013;10:738-46
10. Zhou L, Wang H, Jing J, Yu L, Wu X, Lu Z. Regulation of hair follicle development by exosomes derived from dermal papilla cells. Biochemical and Biophysical Research Communications. 2018;500:325-32
11. Zhou L, Wang H, Jing J, Yu L, Wu X, Lu Z. Regulation of hair follicle development by exosomes derived from dermal papilla cells. Biochemical and biophysical research communications. 2018
12. Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nature cell biology. 2007;9:654
13. Yuan A, Farber EL, Rapoport AL, Tejada D, Deniskin R, Akhmedov NB. et al. Transfer of microRNAs by embryonic stem cell microvesicles. PloS one. 2009;4:e4722
14. Lässer C, Alikhani VS, Ekström K, Eldh M, Paredes PT, Bossios A. et al. Human saliva, plasma and breast milk exosomes contain RNA: uptake by macrophages. Journal of translational medicine. 2011;9:9
15. Xu J-F, Yang G-h, Pan X-H, Zhang S-J, Zhao C, Qiu B-S. et al. Altered microRNA expression profile in exosomes during osteogenic differentiation of human bone marrow-derived mesenchymal stem cells. PloS one. 2014;9:e114627
16. Mardaryev AN, Ahmed MI, Vlahov NV, Fessing MY, Gill JH, Sharov AA. et al. Micro-RNA-31 controls hair cycle-associated changes in gene expression programs of the skin and hair follicle. The FASEB Journal. 2010;24:3869-81
17. Ahmed MI, Alam M, Emelianov VU, Poterlowicz K, Patel A, Sharov AA. et al. MicroRNA-214 controls skin and hair follicle development by modulating the activity of the Wnt pathway. J Cell Biol. 2014;207:549-67
18. Gao Y, Wang X, Yan H, Zeng J, Ma S, Niu Y. et al. Comparative Transcriptome Analysis of Fetal Skin Reveals Key Genes Related to Hair Follicle Morphogenesis in Cashmere Goats. PLOS ONE. 2016:11
19. Yuan C, Wang X, Geng R, He X, Qu L, Chen Y. Discovery of cashmere goat (Capra hircus) microRNAs in skin and hair follicles by Solexa sequencing. BMC genomics. 2013;14:511
20. Zhou G, Kang D, Ma S, Wang X, Gao Y, Yang Y. et al. Integrative analysis reveals ncRNA-mediated molecular regulatory network driving secondary hair follicle regression in cashmere goats. BMC genomics. 2018;19:222
21. Harrison ML, Gore ME, Spriggs D, Kaye S, Iasonos A, Hensley M. et al. Downregulation of Endothelial MicroRNA-200b Supports Cutaneous Wound Angiogenesis By Desilencing GATA Binding Protein 2 and Vascular Endothelial Growth Factor Receptor 2. Arterioscler Thromb Vasc Biol. 2012;32:1372-82
22. Aunin E, Broadley D, Ahmed MI, Mardaryev AN, Botchkareva NV. Exploring a Role for Regulatory miRNAs In Wound Healing during Ageing:Involvement of miR-200c in wound repair. Scientific Reports. 2017:7
23. He N, Dong Z, Li T, Zhao S, Bou S, Liu D. Isolation and characterization of hair follicle stem cells from Arbas Cashmere goat. Cytotechnology. 2016;68:1-10
24. Bickenbach JR, Chism E. Selection and extended growth of murine epidermal stem cells in culture. Experimental Cell Research. 1998;244:184-95
25. He X, Chao Y, Zhou G, Chen Y. Fibroblast growth factor 5-short (FGF5s) inhibits the activity of FGF5 in primary and secondary hair follicle dermal papilla cells of cashmere goats. Gene. 2016;575:S0378111915011439
26. Zhou G, Yuan C, He X, Kang D, Wang X, Chen Y. Effect of miR-125b on dermal papilla cells of goat secondary hair follicle. Electronic Journal of Biotechnology. 2016;25:64-9
27. Zhu B, Xu T, Yuan J, Guo X, Liu D. Transcriptome sequencing reveals differences between primary and secondary hair follicle-derived dermal papilla cells of the Cashmere goat (Capra hircus). PloS one. 2013;8:e76282
28. Title AC, Denzler R, Stoffel M. Uptake and Function Studies of Maternal Milk-derived MicroRNAs*. Journal of Biological Chemistry. 2015;290:23680-91
29. Théry C, Amigorena S, Raposo G, Clayton A. Isolation and Characterization of Exosomes from Cell Culture Supernatants and Biological Fluids: John Wiley & Sons, Inc. 2006.
30. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet Journal. 2011:17
31. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform: Oxford University Press. 2009.
32. Zhou L, Chen J, Li Z, Li X, Hu X, Huang Y. et al. Integrated profiling of microRNAs and mRNAs: microRNAs located on Xq27.3 associate with clear cell renal cell carcinoma. PloS one. 2010;5:e15224
33. Storey JD. The positive false discovery rate: a Bayesian interpretation and the q-value. Annals of Statistics. 2003;2003:2013-35
34. KC M, T H, Y T, YS A, WL T, AM T. et al. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203-17
35. Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nature Genetics. 2007;39:1278-84
36. Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biology. 2010;11:R14-R
37. Mao XZ, Cai T, Olyarchuk JG, Wei LP. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005;21:3787-93
38. Ito M, Liu Y, Yang Z, Nguyen J, Liang F, Morris RJ. et al. Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nature medicine. 2005;11:1351-4
39. Tausche AK, Skaria M, Böhlen L, Liebold K, Hafner J, Friedlein H. et al. An autologous epidermal equivalent tissue-engineered from follicular outer root sheath keratinocytes is as effective as split-thickness skin autograft in recalcitrant vascular leg ulcers. Wound Repair & Regeneration. 2010;11:248-52
40. Shim JH. Hair growth-promoting effect of human dermal stem/progenitor cell-derived conditioned medium. Tissue Engineering & Regenerative Medicine. 2015;12:268-75
41. Rajendran RL, Gangadaran P, Bak SS, Oh JM, Kalimuthu S, Lee HW. et al. Extracellular vesicles derived from MSCs activates dermal papilla cell in vitro and promotes hair follicle conversion from telogen to anagen in mice. Sci Rep. 2017;7:15560
42. Cotsarelis G, Sun T-T, Lavker RM. Label-retaining cells reside in the bulge area of pilosebaceous unit: implications for follicular stem cells, hair cycle, and skin carcinogenesis. Cell. 1990;61:1329-37
43. Ito M, Cotsarelis G, Kizawa K, Hamada K. Hair follicle stem cells in the lower bulge form the secondary germ, a biochemically distinct but functionally equivalent progenitor cell population, at the termination of catagen. Differentiation. 2010;72:548-57
44. Lei M, Yang L, Chuong CM. Getting to the Core of the Dermal Papilla. Journal of Investigative Dermatology. 2017;137:2250-3
45. Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654-9
46. Liang Y, Ridzon D, Wong L, Chen C. Characterization of microRNA expression profiles in normal human tissues. BMC genomics. 2007;8:166
47. Lien WH, Guo X, Polak L, Lawton LN, Young RA, Zheng D. et al. Genome-wide maps of histone modifications unwind in vivo chromatin states of the hair follicle lineage. Cell stem cell. 2011;9:219-32
48. Greco V, Chen T, Rendl M, Schober M, Pasolli HA, Stokes N. et al. A Two-Step Mechanism for Stem Cell Activation during Hair Regeneration. Cell stem cell. 2009;4:155-69
49. Hsu YC, Li L, Fuchs E. Emerging interactions between skin stem cells and their niches. Nature medicine. 2014;20:847-56
50. Kandyba E, Leung Y, Chen YB, Widelitz R, Chuong CM, Kobielak K. Competitive balance of intrabulge BMP/Wnt signaling reveals a robust gene network ruling stem cell homeostasis and cyclic activation. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:1351-6
51. Lay K, Kume T, Fuchs E. FOXC1 maintains the hair follicle stem cell niche and governs stem cell quiescence to preserve long-term tissue-regenerating potential. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:E1506
52. Lim X, Tan SH, Yu KL, Lim SB, Nusse R. Axin2 marks quiescent hair follicle bulge stem cells that are maintained by autocrine Wnt/β-catenin signaling. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:201601599
53. Ma X, Tian Y, Song Y, Shi J, Xu J, Xiong K. et al. Msi2 maintains quiescent state of hair follicle stem cells by directly repressing the Hh signaling pathway. Journal of Investigative Dermatology. 2017;137:1015
54. Yuan S, Li F, Meng Q, Zhao Y, Chen L, Zhang H. et al. Post-transcriptional Regulation of Keratinocyte Progenitor Cell Expansion, Differentiation and Hair Follicle Regression by miR-22. PLoS Genet. 2015;11:e1005253
55. Paus R, Foitzik K. In search of the “hair cycle clock”: a guided tour. Differentiation. 2010;72:489-511
56. Ophelia V, Tetsuro K, Yoichi I, Wado A, Takashi S, Shinya Y. et al. Human induced pluripotent stem cell-derived ectodermal precursor cells contribute to hair follicle morphogenesis in vivo. Journal of Investigative Dermatology. 2013;133:1479-88
57. Baker RE, Murray PJ. Understanding hair follicle cycling: a systems approach. Current opinion in genetics & development. 2012;22:607-12
58. Kishimoto J, Burgeson RE, Morgan BA. Wnt signaling maintains the hair-inducing activity of the dermal papilla. Genes & development. 2000;14:1181-5
59. Kandyba E, Kobielak K. Wnt7b is an important intrinsic regulator of hair follicle stem cell homeostasis and hair follicle cycling. Stem cells. 2014;32:886-901
60. Udayan G, Lars M, Pamela C, Lixin K, W Michael OG, Dolores DV. et al. Bone morphogenetic protein signaling regulates postnatal hair follicle differentiation and cycling. American Journal of Pathology. 2004;165:729-40
61. Won CH, Kwon OS, Kang YJ, Yoo HG, Lee DH, Chung JH. et al. Comparative secretome analysis of human follicular dermal papilla cells and fibroblasts using shotgun proteomics. Bmb Reports. 2012;45:253
62. C Tr, S A, G R, A C. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006;30:Unit 3.22
63. Rajendran RL, Gangadaran P, Bak SS, Ji MO, Kalimuthu S, Lee HW. et al. Extracellular vesicles derived from MSCs activates dermal papilla cell in vitro and promotes hair follicle conversion from telogen to anagen in mice. Sci Rep. 2017;7:15560
64. Tadokoro H, Umezu T, Ohyashiki K, Hirano T, Ohyashiki JH. Exosomes derived from hypoxic leukemia cells enhance tube formation in endothelial cells. The Journal of biological chemistry. 2013;288:34343-51
65. Yu F, Yusuke Y, Takahiro O. Extracellular vesicle transfer of cancer pathogenic components. Cancer Science. 2016;107:385-90
66. Du KT, Deng JQ, He XG, Liu ZP, Peng C, Zhang MS. MiR-214 Regulates the Human Hair Follicle Stem Cell Proliferation and Differentiation by Targeting EZH2 and Wnt/β-Catenin Signaling Way In Vitro. Tissue Engineering & Regenerative Medicine. 2018:1-10
67. Adam RC, Yang H, Rockowitz S, Larsen SB, Nikolova M, Oristian DS. et al. Pioneer factors govern super-enhancer dynamics in stem cell plasticity and lineage choice. Nature. 2015;521:366
68. Sennett R, Wang Z, Rezza A, Grisanti L, Roitershtein N, Sicchio C. et al. An Integrated Transcriptome Atlas of Embryonic Hair Follicle Progenitors, their Niche and the Developing Skin. Developmental cell. 2015;34:577-91
69. Chang H, Cahill H, Smallwood PM, Wang Y, Nathans J. Identification of Astrotactin2 as a Genetic Modifier That Regulates the Global Orientation of Mammalian Hair Follicles. Plos Genetics. 2015;11:e1005532
70. Schmuth M, Jiang YJ, Dubrac S, Elias PM, Feingold KR. Thematic review series: skin lipids. Peroxisome proliferator-activated receptors and liver X receptors in epidermal biology. Journal of lipid research. 2008;49:499-509
71. Ludwig AK, Giebel B. Exosomes: Small vesicles participating in intercellular communication. International Journal of Biochemistry & Cell Biology. 2012;44:11-5
72. Helen S, Emily H, Helen T, Rebecca D, Peter T, Chern Ein O. et al. New mechanism for Notch signaling to endothelium at a distance by Delta-like 4 incorporation into exosomes. Blood. 2010;116:2385
73. Tarutani M, Itami S, Okabe M, Ikawa M, Tezuka T, Yoshikawa K. et al. Tissue-specific knockout of the mouse Pig-a gene reveals important roles for GPI-anchored proteins in skin development. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:7400-5
74. Azoulay-Alfaguter I, Mor A. Proteomic analysis of human T cell-derived exosomes reveals differential RAS/MAPK signaling. Eur J Immunol. 2018;48:1915-7
75. Botchkarev VA, Kishimoto J. Molecular Control of Epithelial-Mesenchymal Interactions During Hair Follicle Cycling. The journal of investigative dermatology Symposium proceedings. 2003;8:46-55
76. Roh C, Tao Q, Lyle S. Dermal papilla-induced hair differentiation of adult epithelial stem cells from human skin. Physiological Genomics. 2004;19:207
77. Millar SE. Molecular Mechanisms Regulating Hair Follicle Development. Journal of Investigative Dermatology. 2002;118:216-25
78. Millar SE, Willert K, Salinas PC, Roelink H, Nusse R, Sussman DJ. et al. WNT Signaling in the Control of Hair Growth and Structure ☆. Developmental Biology. 1999;207:133-49
79. C vG, RM O, I F, RG Q, TG P, L B. et al. Development of several organs that require inductive epithelial-mesenchymal interactions is impaired in LEF-1-deficient mice. Genes & development. 1994;8:2691-703
80. Zhou P, Byrne C, Jacobs J, Fuchs E. Lymphoid enhancer factor 1 directs hair follicle patterning and epithelial cell fate. Genes & development. 1995;9:700-13
Author contact
![]() Corresponding authors: College of Animal Science and Technology, Northwest A&F University, Yangling, Shaanxi, China; E-mail: chenyulinedu.cn (Yulin Chen); zengwenxian2015com (Wenxian Zeng)
Corresponding authors: College of Animal Science and Technology, Northwest A&F University, Yangling, Shaanxi, China; E-mail: chenyulinedu.cn (Yulin Chen); zengwenxian2015com (Wenxian Zeng)