Impact Factor ISSN: 1449-2288
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(11):4560-4577. doi:10.7150/ijbs.69933 This issue Cite
Research Paper
TCF7L2 promotes anoikis resistance and metastasis of gastric cancer by transcriptionally activating PLAUR
Tao Zhang1,2,3*, Bofang Wang2,3*, Fei Su1*, Baohong Gu2,3*, Lin Xiang2,3, Lei Gao2,3, Peng Zheng2,3, Xue-mei Li2,3, Hao Chen2,3,4 ![]()
1. Department of oncology, The First Hospital of Lanzhou University, Lanzhou, Gansu, China.
2. The second clinical medical college of Lanzhou university, Lanzhou , Gansu, China.
3. Key laboratory of digestive system tumors, Lanzhou University Second Hospital, Lanzhou, Gansu, China.
4. Cancer center, Lanzhou University Second Hospital, Lanzhou, Gansu, China.
*These authors contributed equally to this work.
Received 2021-12-10; Accepted 2022-6-14; Published 2022-7-11
Abstract

Gastric cancer (GC) is the most common gastrointestinal malignant tumor, and distant metastasis is a critical factor in the prognosis of patients with GC. Understanding the mechanism of GC metastasis will help improve patient prognosis. Studies have confirmed that urokinase-type plasminogen activator receptor (PLAUR) promotes GC metastasis; however, its relationship with anoikis resistance and associated mechanisms remains unclear. In this study, we demonstrated that PLAUR promotes the anoikis resistance and metastasis of GC cells and identified transcription Factor 7 Like 2 (TCF7L2) as an important transcriptional regulator of PLAUR. We also revealed that TCF7L2 is highly expressed in GC and promotes the anoikis resistance and metastasis of GC cells. Moreover, we found that TCF7L2 transcription activates PLAUR. Finally, we confirmed that TCF7L2 is an independent risk factor for poor prognosis of patients with GC. Our results show that TCF7L2 and PLAUR are candidate targets for developing therapeutic strategies for GC metastasis.
Keywords: gastric cancer, urokinase-type plasminogen activator receptor, transcription Factor 7 Like 2, transcription, anoikis
Introduction
Gastric cancer (GC) is the most common malignant tumor of the digestive system and the fifth most commonly diagnosed cancer worldwide [1]. Compared to European and American countries, the morbidity and mortality of GC are higher in China [2]. Despite the significantly improved prognosis of patients with GC with the development of novel drugs and advancement in therapeutic strategies, the long-term survival of patients with advanced GC remains dire. Distant metastasis is the primary influencing factor in the prognosis of patients with GC [3]. Hence, it is necessary to understand the mechanism of GC metastasis and to identify metastasis-related molecular markers and therapeutic targets.
“Anoikis” is cell death that occurs when cells lose interaction with surrounding stromal cells [6]. Anoikis resistance is a hallmark event in the migration of cancer cells, as they must be able to survive after detachment from their supporting cells [4, 5]. In cancer cells, anoikis resistance is regulated by multiple genes and signaling pathways [7, 8]. Anoikis resistance plays a significant role in GC metastasis (Figure 1). For example, the nuclear MYH9-induced CTNNB1 enhances the anoikis resistance of GC cells, ultimately promoting peritoneal metastasis of GC [9]. L1CAM is upregulated in GC and is closely associated with the anoikis resistance of tumor cells [10]. Briefly, cancer cells must overcome anoikis for successful metastasis.
Relationship between anoikis resistance and metastasis of cancer cells.
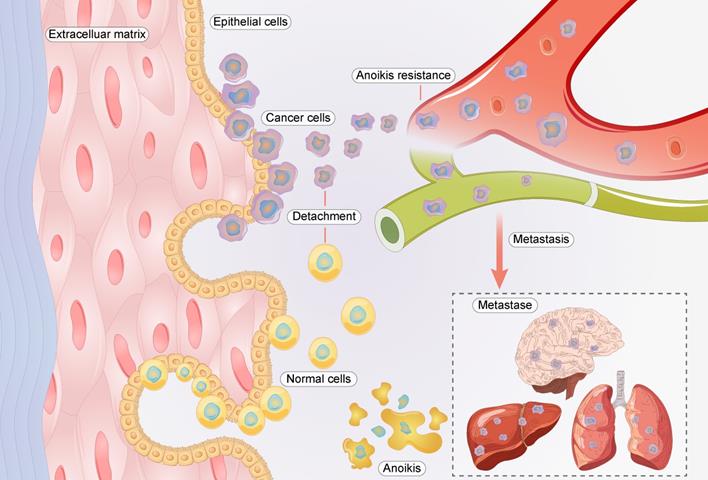
As cancer malignancy progresses, the urokinase-type plasminogen activator receptor (uPAR or PLAUR) plays a key role, as it is not expressed in normal tissues but is strongly expressed in tumor tissues. The expression of PLAUR is linked to the invasion and migration of tumor cells [11-13]. In tumor tissues, PLAUR can activate extracellular matrix proteolytic enzymes outside the cells by binding to urokinase-type plasminogen activator (uPA or PLAU), its ligand. This binding causes pathological changes in the extracellular matrix, ultimately creating favorable conditions for malignant behaviors such as the invasion and migration of tumor cells [14, 15]. Numerous studies have demonstrated that PLAUR is upregulated in GC and is closely associated with cancer cell invasion and migration [16-18]. However, its effect on the anoikis resistance of GC cells and its mechanisms remain unclear.
Transcription Factor 7 Like 2 (TCF7L2) is a member of the high-mobility group (HMG) family, a downstream effector of the Wnt/β-catenin signaling pathway, and a pivotal regulator of development and cell growth [19-22]. TCF7L2 has a crucial role in various biological processes and functions of numerous organs and tissues, including liver, pancreatic islets, and adipose tissues [23, 24]. Studies have suggested the close correlation of TCF7L2 with various somatic metabolic disorders. TCF7L2 leads to type 2 diabetes, primarily by affecting the metabolism and homeostasis of glucose [20, 25, 26]. Recent studies have also confirmed its strong linkage to cancer. For example, the TCF7L2 gene polymorphism is closely associated with the onset of familial breast, colorectal, prostatic, and esophageal cancers [27-31]. However, the correlation of TCF7L2 with GC as well as its effect on the anoikis resistance of GC cells has rarely been reported.
This study explored the association of PLAUR with the anoikis resistance of GC cells. TCF7L2 was identified as a transcriptional regulator of PLAUR. We confirmed that TCF7L2 activated the classic caspase-3/7 pathway in a PLAUR-dependent manner, thereby enhancing the anoikis resistance and migration of GC cells.
Materials and Methods
Clinical samples
Paired GC tissues and adjacent non-cancer tissues were acquired from the Lanzhou University Second Hospital. Cohort 1: PLAUR mRNA expression was detected in 15 pairs of fresh GC tissues and adjacent tissues, and the correlation between PLAUR and TCF7L2 protein expression was analyzed via immunofluorescence (IF) in these 15 GC tissues. Cohort 2: 121 pairs of GC tissues and adjacent non-cancer tissues were formalin-fixed and paraffin-embedded and then used to detect the expression of TCF7L2 protein in GC and adjacent tissues by immunohistochemistry (IHC). Cohort 3: 10 pairs of fresh GC tissues and adjacent non-cancer tissues were cryopreserved and used for quantitative analysis of TCF7L2 protein expression in GC and adjacent tissues by western blot. All enrolled patients signed informed consent, and procedures were approved by the Ethics Committee of Lanzhou University Second Hospital.
Cell culture
GC cell lines MKN45, MGC-803, AGS, and HGC-27; gastric mucosal normal epithelial cell GES-1; and human embryonic kidney 293T (HEK293T) cells were purchased from Shanghai Fuheng Biotechnology (Shanghai, China). All cell lines were cultivated in Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin streptomycin (Beyotime, Shanghai, China) in a 37 °C incubator with 5% CO2 under humidifying conditions.
Cell transfection
Small hairpin RNA (shRNA) targeting PLAUR and TCF7L2, and negative control shRNA (sh-Ctrl) were synthesized by Compass Biotech (Quanzhou, China). The coding sequence of PLAUR or TCF7L2 was amplified and cloned into pCDH-CMV-MCS-EF1-copGFP plasmid (Compass Biotech) and packaged as lentivirus. Lentivirus was transfected into candidate GC cell lines. Positive cells were screened with puromycin (2 μg /mL) for 2 weeks, and the effect of knockdown and overexpression efficiency was measured via quantitative reverse transcription polymerase chain reaction (qRT-PCR) and western blot. The target sequences of PLAUR and TCF7L2 shRNAs are provided in Table S1.
Construction of anoikis model
For the construction of the anoikis model, 1×106 cells were suspended in ultra-low adsorption 6-well plates (Corning, NY, USA) and cultured for 24 h. Suspended cells were harvested and processed for subsequent experiments.
qRT-PCR
Trizol reagent (Invitrogen, Grand Island, NY, USA) was used to extract total RNA from GC cells and tissue samples, according to the manufacturer's protocols. Total RNA was used for reverse transcription using PrimeScriptTM RT Reagent Kit (Takara, Shiga, Japan). Quantification was performed with SYBR Green PCR Master Mix (Takara) using Applied Biosystems ABI 7500 (Applied Biosystems, Waltham, MA, USA). GAPDH was used as an internal reference gene. The 2-ΔΔCt method was utilized to quantify fold changes. All experiments were repeated in triplicate. Primer sequences are provided in Table S2.
Western blot
Radio immunoprecipitation assay (RIPA, Beyotime, Shanghai, China) supplemented with protease inhibitors (Beyotime) was used to extract total protein from GC cells and tissue samples. Total protein was run on 8-12% SDS/PAGE gels and transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Boston, MA, USA). Subsequently, membranes were blocked with 5% skim milk and incubated overnight at 4 °C with the primary antibodies. The next day, PVDF membranes were incubated for 2 h at room temperature with appropriate secondary antibodies, and ECL substrate was applied. Images were visualized using a Tianneng automatic chemiluminescence image processing system (TANO, Shanghai, China). Primary antibodies are presented in Table S3.
Cell proliferation assay
The cell counting kit 8 (CCK-8, Yelasen, Shanghai, China) was used to quantify the proliferation ability of GC cells, according to the manufacturer's instructions. Cells in the sh-Ctrl group and shRNA group were seeded in 96-well plates (Corning) at 1×104 cells/well. After 24 h, 48 h, and 72 h, 10 μL of CCK8 solution was mixed with the cells and incubated for 2 h at 37 °C to assess proliferation activity. The absorbance was measured at 450 nm using a BioTek microplate reader (BioTek, Winooski, VT).
5-ethynyl-2'-deoxyuridine (EdU) assay
An EdU assay was performed using an EdU assay kit (RiboBio, Guangzhou, China). Cells were incubated with 50 μm Edu solution at room temperature for 6-8 h. Subsequently, cells were fixed with 4% neutral paraformaldehyde at room temperature for 30 min. Cells were permeabilized with PBS containing 0.5% Triton X-100 for 20 min and then washed three times with PBS. Cells were stained with Apollo staining reagent (KeyGEN, Nanjing, China) for 20 min and washed three times with NaCl/PI. Cell nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI, KeyGEN) and visualized under a microscope.
Transwell assay
Transwell chambers (Corning) were used to measure cell migration ability. Cells were suspended in RPMI 1640 medium without FBS, and 1×104 cells were seeded into the upper chamber of the transwell chamber. Subsequently, 800 μL of RPMI 1640 medium with 10% FBS was added into the lower chamber. After 24 h, cells that had migrated to the lower surface of membranes were fixed with 4% paraformaldehyde for 20 min and stained with 0.1% crystal violet for 20 min. The average number of migrating cells was determined by six randomly picked fields of vision under the microscope.
Wound healing assay
The wound healing assay was processed using 6-well plates (Corning). The cells of different groups were seeded into 6-well plates and cultivated until the cells were 90% confluent. A scratch was made with a pipettor tip, the detached cells were washed off with PBS, and then cells were cultured in medium without FBS for 24 h. The wound widths were assessed at 0 h and 24 h under a light microscope, and ImageJ software (ImageJ Software Inc., MD, USA) was used to assess the relative area of wound closure.
Cell apoptosis assay
Flow cytometry was used for apoptosis assays. The AnnexinV-FITC apoptosis detection kit (Yelasen, Shanghai, China) was used to measure the apoptosis rates of adherent and suspension cells. Cells were washed twice with PBS buffer, 300 μL of binding buffer was added to tubes, and stained by 5 μL Annexin V-FITC and 10 μL PI in the dark for 15 min at room temperature. The percentage of cell apoptosis was measured via flow cytometry (Beckman Cytoflex, California, CA, USA).
IF
Cells or tissues on slides were fixed with paraformaldehyde (4%) and permeabilized with 0.01% Triton X-100 for 15 min. The slides were incubated with primary antibodies anti-PLAUR (1:200, Santa Cruz, sc-13522) or anti-TCF7L2 (1:100, Bioss, bsm-52543R), overnight in a humidified chamber at 37 °C in the dark. Subsequently, cell nuclei were stained with DAPI. The fluorescence distribution and intensity of cells and tissues were analyzed using a Zeiss LSM 880 laser microscope (Carl Zeiss AG, Oberkochen, Germany).
DNA pull-down assay
The DNA-pull-down assay was performed using the Bes5004 DNA pull-down kit (BersinBio, Guangzhou, China), according to the manufacturer's instructions. First, the PLAUR promoter sequence was searched for in the NCBI (Table S4). PCR products were labeled with biotin using a universal biotinylated primer. The gel recovery kit (QIAGEN, Venlo, Netherlands) was used to isolate a labeled probe from agarose gels. Lysis buffer was used to extract MKN45 and 293T cell total protein, and a BCA kit (Solarbio, Beijing, China) was used to measure the protein concentration. One part of the protein was marked as “Input,” and the other part was incubated with the probe to form a protein-DNA-magnetic bead complex. The targeted protein was marked as “Target” after it was eluted and then run on SDS-PAGE. Differential bands were analyzed by silver staining. The fractions were analyzed using nanospray liquid chromatography tandem mass spectrometry (LC-MS/MS) on a Thermo Scientific QE HF (Thermo Fisher Scientific, MA, USA).
Chromatin-immunoprecipitation (ChIP)
ChIP assays were executed using the SimpleChIP® Enzymatic Chromatin IP Kit (CST, Massachusetts, MA, USA). The cells were fixed by 1% paraformaldehyde for 10 min, then 0.125 M glycine was added and the mixture was placed at room temperature for 5 min to terminate the DNA-protein crosslinking. SDS lysis buffer (with protease inhibitors) was added to the cells, and an ultrasonic fragmentation device was used to generate chromatin fragments. A portion of the products was marked as “Input.” The remaining lysates were incubated with TCF7L2 antibody (Abcam, Cambridge, UK) and Protein G magnetic beads to form a DNA-antibody-magnetic beads complex, and the DNA was marked as “Target” after elution and purification. Rabbit IgG (CST, Massachusetts, MA, USA) served as a negative control. The final purified DNA fragment was analyzed via qRT-PCR. Primers of qPCR for the PLAUR promoter binding site are presented in Table S5.
Luciferase activity assay
Luciferase activity assay was performed using the Luciferase Detection Kit (Promega, Madison, WI, USA), according to the manufacturer's instruction. First, The WT and mutated PLAUR UTR were cloned into PGL4.11-Basic Vector (Promega), producing the vectors PGL4.11-PLAUR-WT-promoter-luc and PGL4.11-PLAUR-mut-promoter-luc MKN45, and 293T cells were co-transfected with different plasmids for 48 h. Finally, the Promega dual-luciferase system (Promega) was used for detection.
Co-immunoprecipitation (co-IP) assay
Co-IP assay was performed as described previously [32]. Lysates were incubated with Flag affinity beads (Sigma-Aldrich, MA, USA). The interacting proteins were detected via western blot.
Xenograft assay
MKN45 cells were transfected with shRNA against PLAUR, TCF7L2 and the corresponding scramble interference by lentiviral infection. Each plasmid had a luciferase label. Cells transfected with sh-Ctrl and shRNAs (sh-PLAUR or sh-TCF7L2) 1:1 were mixed with PBS and Matrigel suspension reagent (BD Biosystems, San Jose, CA, USA). Briefly, 1×106 cells were injected subcutaneously into female BALB/c nude mice aged 6-8 weeks (Hangzhou Ziyuan Laboratory Animal Science and Technology). Tumor volumes were measured every 4 days and calculated by the formula (length × width2)/2. After 24 days, mice were sacrificed by deep anesthesia with ethyl ether. Tumor tissues were harvested for subsequent experiments. The lung metastasis xenograft model was established by injecting 1×106 cells into female BALB/c nude mice aged 6-8 weeks through the tail vein. The bioluminescence images were acquired by the IVIS spectrum imaging system (PerkinElmer, Massachusetts, USA). Lastly, mice were sacrificed after 28 days, and lung tissues were harvested for subsequent experiments. Animal experiments were reviewed and approved by the Animal Use Ethics Committee of Lanzhou University Second Hospital. All animals were cared for in accordance with the Guide for the “Care and Use of Laboratory Animals”.
Hematoxylin and eosin (H&E) staining and IHC
H&E staining was performed on 5 μm paraffin sections using a standard H&E staining protocol [33]. Tissue samples were fixed, embedded, cut into slices, dewaxed, antigen retrieved, incubated with primary antibody, incubated with secondary antibody, DAB stained, hematoxylin stained, and alcohol denatured. The primary antibodies were anti-PLAUR (1:200, Santa Cruz, sc-13522), anti-TCF7L2 (1:100, CST, #2569), and anti-Ki67 (1:100, Proteintech, 27309-1-AP). Two experienced pathologists independently reviewed all sections. The IHC score was determined using modified Histo-score (H-score) [34]. The staining intensity and percentage of positive cells were scored as described previously [35]. The final immunoreactive score was calculated as the staining intensity score × the percentage of positive cells. The expression level of TCF7L2 was considered “low expression” if the immunoreactive score was 0-4 and “high expression” if the score was 5-9.
Bioinformatics analyses
Stomach adenocarcinoma (STAD) gene mutation data and RNA-seq data were obtained from the TCGA database. GenVisR [36] data package was used to construct and draw the “waterfall map” of STAD gene mutation. UCSC Genome Browse (http://genome.ucsc.edu) [37] and PROMO database (http://alggen.lsi.upc.es/home.html) [38] were used to search for PLAUR promoter sequences and analyze transcription factors that regulate PLAUR. The Pearson correlation between the expression of PLAUR and its transcription factor TCF7L2 was calculated using the “ggstatsplot” and “ggplot2” packages. The JASPAR database (https://jaspar.genereg.net/) [39] was used to identify the binding site of transcription factor TCF7L2 to the PLAUR promoter. Expression profiles and protein level of TCF7L2 in STAD were acquired from GEPIA (http://gepia.cancer-pku.cn/) [40] and HPA (https://www.proteinatlas.org) [41] databases. The survival of patients with GC against TCF7L2 expression was estimated using a Kaplan-Meier plotter (http://www.kmplot.com/) [42].
Statistical analysis
Functional analysis was performed using the SPSS software package (version 24.0, IBM SPSS, IL, USA) and GraphPad Prism (version 8.0, GraphPad Software, CA, USA). We used Student's t-test to examine the difference in means between two groups, and two-tailed ANOVA to examine the difference between multiple groups. A Chi-square test was used for correlation analysis between gene expression and clinicopathological features. The Kaplan-Meier method was used to test the overall survival between different groups. Cox regression analysis was used to analyze the influencing factors affecting the prognosis of patients. A P value < 0.05 was considered a statistically significant difference.
Results
PLAUR promotes the proliferation, anoikis resistance, and migration of GC cells
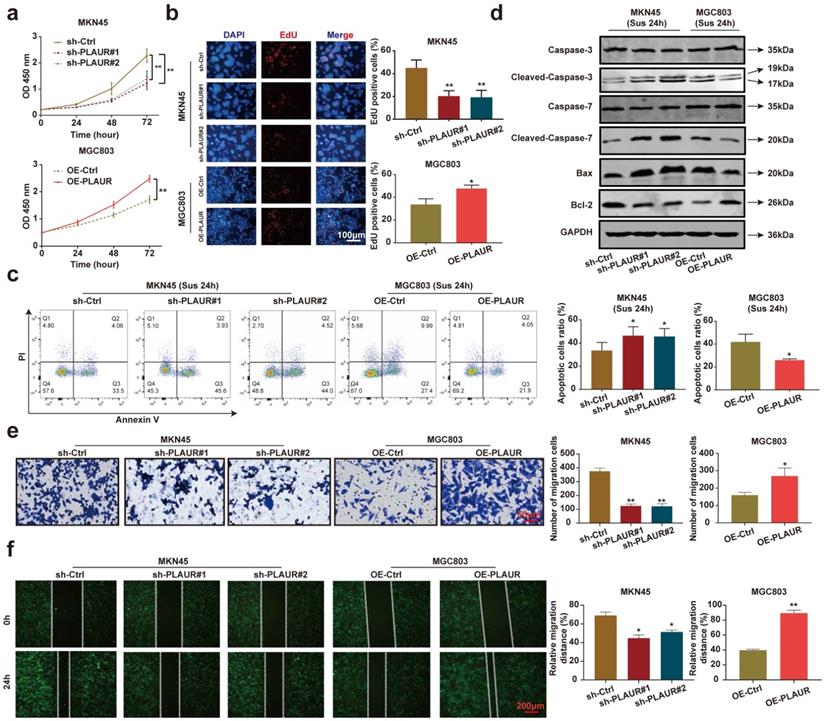
To analyze the effects of PLAUR on the malignant behavior of GC cells, we quantified its expression in normal gastric epithelial cells (GES-1) and GC cell lines (HGC27, MGC803, AGS, and MKN45). The expression of PLAUR was highest in MKN45 cells and lowest in MGC803 cells (Supplementary Figure 1). Accordingly, these two cell lines were used to construct stable PLAUR knockdown or overexpressing cells. qRT-PCR and western blot were used to confirm the knockdown or overexpression (Supplementary Figures 2). Afterwards, we assayed the effects of PLAUR on GC cell proliferation via CCK-8 and EdU assays. The results showed that the knockdown of PLAUR inhibited the proliferation of MKN45 cells, while its overexpression promoted the proliferation of MGC803 cells (Figure 2a-b). To mimic the anoikis of GC cells, we used ultra-low attachment 6-well plates to suspend the cells for 24 h. Thereafter, the impact of PLAUR on the anoikis resistance of GC cells was analyzed via flow cytometry. Following a 24 h suspension of cells, the knockdown of PLAUR promoted the apoptosis of MKN45 cells, whereas its overexpression inhibited the apoptosis of MGC803 cells (Figure 2c). Meanwhile, we used western blot to detect the apoptosis-related protein expression in the GC cells that had been suspended for 24 h. As shown in Figure 2d, after cell suspension for 24 h, the knockdown of PLAUR promoted the expression of cleaved-caspase-3/7 and Bax in the MKN45 cells, while inhibiting the expression of Bcl-2. In contrast, the overexpression of PLAUR inhibited cleaved-caspase-3/7 and Bax expression in MGC803 cells, while promoting the expression of Bcl-2. Finally, transwell and wound healing assays were employed to analyze the correlation of PLAUR with the migration of GC cells. The knockdown of PLAUR inhibited the migration of MKN45 cells, while its overexpression promoted the migration of MGC803 cells (Figure 2e-f). Overall, PLAUR promoted the proliferation, anoikis resistance, and migration of GC cells.
PLAUR promotes the malignant phenotype of GC cells. a-b Effects of PLAUR knockdown or overexpression on the proliferation of GC cells, measured by CCK-8 (a) and EdU assay (b). c After GC cells were suspended for 24h, the effects of PLAUR knockdown or overexpression on the anoikis resistance of GC cells, analyzed via flow cytometry. d Changes in apoptosis-related proteins after PLAUR knockdown or overexpression were analyzed via western blot in GC cells suspended for 24 h. e-f Effects of PLAUR knockdown or overexpression on the migration of GC cells, evaluated by transwell (e) and wound healing assay (f). Compared with the corresponding control group, * P < 0.05, ** P < 0.001.
PLAUR promotes tumor growth and metastasis in vivo
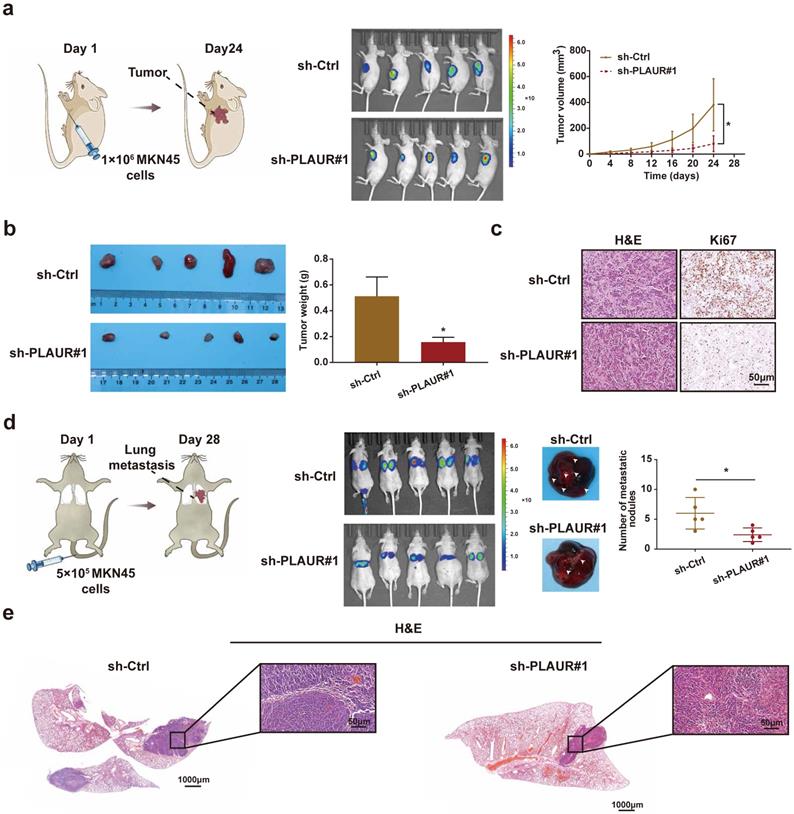
To further confirm our in vitro observations, we explored whether PLAUR regulates the growth and metastasis of tumor cells in vivo. Initially, we established a nude mouse model of subcutaneous tumors by subcutaneously injecting MKN45 cells with sh-Ctrl or sh-PLAUR#1. We observed the changes in subcutaneous tumor volume of nude mice and plotted corresponding growth curves. The tumors were also confirmed by small animal live imaging on the 24th day after injection. As expected, PLAUR knockdown significantly delayed in vivo tumor growth in nude mice (Figure 3a). Tumors harvested from the sacrificed animals were weighed, and significantly heavier tumors were observed in nude mice in the sh-Ctrl group than in the sh-PLAUR#1 group (Figure 3b). Additionally, subcutaneous tumors were identified via H&E staining (Figure 3c). IHC analysis revealed that PLAUR knockdown inhibited the expression of Ki67 in tumor tissues (Figure 3c). Subsequently, we established a nude mouse model of lung metastasis through tail vein injection of MKN45 cells, either with sh-Ctrl or sh-PLAUR#1. On the 28th day following injection, mice in the sh-Ctrl group exhibited significantly higher lung tumor burden than those in the sh-PLAUR#1 group (Figure 3d). After euthanizing and dissecting mice, the PLAUR knockdown reduced the number of nodules on the lung surfaces (Figure 3d). As shown in Figure 3e, H&E staining was also employed to identify lung metastases. Collectively, the above results confirmed that PLAUR facilitates tumor growth and metastasis in vivo.
PLAUR promotes the GC process in vivo. a Knockdown of PLAUR inhibited the growth of subcutaneous xenograft tumors in nude mice. The bioluminescence images of the subcutaneous tumors were displayed in a small animal in vivo imaging system, and the tumor growth curve for 24 days was measured. b Gross specimen and average weight of the tumor after sacrifice. Knockdown of PLAUR inhibited the volume and weight of subcutaneous xenograft tumors in nude mice. c Histological characteristics and Ki67 expression of the tumor were examined via H&E staining and IHC, respectively. The knockdown of PLAUR inhibited the Ki67 expression of subcutaneous xenograft tumors in nude mice. d Knockdown of PLAUR inhibited lung metastasis in nude mice. The bioluminescence image of lung metastases and the count of lung metastatic nodules are displayed, 28 days after tail veins were injected with GC cells. The white arrows represent the lung metastasis nodules. e Lung metastasis tumors were confirmed via H&E staining. Compared with the control group, * P<0.001.
TCF7L2 is a potential transcriptional regulator of PLAUR
As described earlier, PLAUR was upregulated in GC, which promoted the malignancy of tumor cells, including anoikis resistance. Afterwards, we explored the reasons for PLAUR upregulation in GC. Initially, we examined PLAUR mRNA expression in 15 pairs of GC and adjacent tissues (cohort 1). The results indicated considerably higher PLAUR mRNA content in the GC tissues than in the adjacent tissues (Supplementary Figure 3). Additionally, we ruled out the possibility of PLAUR upregulation in GC caused by PLAUR mutations using a bioinformatics approach (Supplementary Figure 4). Meanwhile, considering the major role of abnormal transcriptional regulation in carcinogenesis and development, we speculated that abnormal transcription leads to upregulated PLAUR expression in GC.
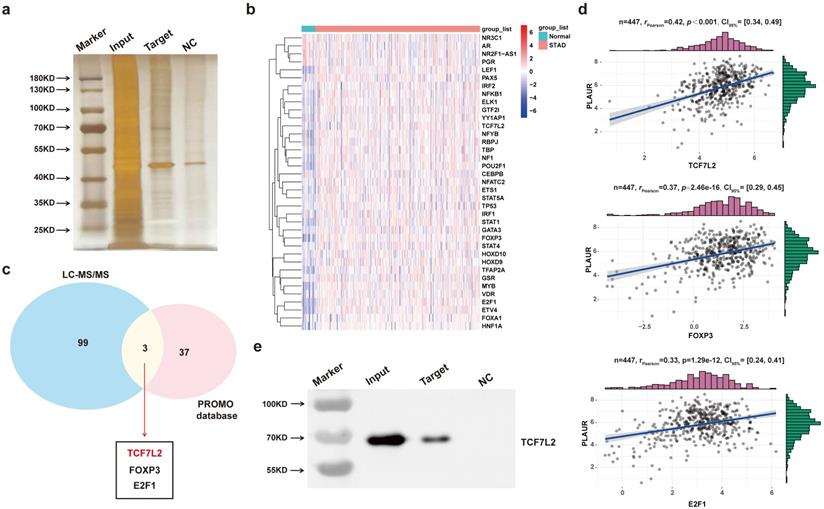
To find major transcription factors capable of regulating PLAUR expression, we first determined the proteins that bind to the PLAUR promoter in MKN45 cells, by DNA pull-down assay. As shown in Figure 4a, many DNA-protein complex bands were found on silver-stained polyacrylamide gels, some of which were absent in the control reaction. We further identified the aforementioned proteins through LC-MS/MS (Table S6). Subsequently, PROMO program was used to discover the potential transcription factors that can regulate PLAUR expression, and TCGA-STAD-seq data were utilized to draw an expression heatmap of these transcription factors in GC (Figure 4b). Notably, three transcription factors, namely TCF7L2, FOXP3, and E2F1, overlapped in the LC-MS/MS and PROMO searches (Figure 4c). Based on the TCGA-STAD-seq data, we analyzed the correlation of PLAUR expression with these transcription factors in GC. As shown in Figure 4d, TCF7L2 exhibited the highest correlation with PLAUR expression (r = 0.42, P < 0.001). Finally, the expression of TCF7L2 protein in the pull-down product was detected via western blot (Figure 4e). Further analysis, aided by online bioinformatics tools, revealed that TCF7L2 was highly expressed in GC, localized primarily in cell nuclei, and was associated with poor patient prognosis (Supplementary Figure 5). Our data suggested that the transcription factor TCF7L2 plays an oncogenic role in GC. Hence, we focused on exploring the regulatory relationship between TCF7L2 and PLAUR.
TCF7L2 is a potential transcriptional regulator of PLAUR. a Silver staining of PLAUR binding protein identified in DNA pulldown. b Heat map of expression of transcription factors of PLAUR in GC samples and normal samples. c TCF7L2, FOXP3, and E2F1 were identified as transcription factors of PLAUR through LC-MS/MS, combined with the screening results of the PROMO database. d Expression correlations of TCF7L2, FOXP3, E2F1, and PLAUR were analyzed based on TCGA-seq data. TCF7L2 has the highest correlation with PLAUR expression. e TCF7L2 protein was confirmed to be present in the pull-down product by western blot.
TCF7L2 transcriptionally upregulates PLAUR expression in GC
To further assess the regulation of PLAUR expression by TCF7L2 at the transcriptional level, we studied the localization of TCF7L2 and PLAUR proteins in MKN45 cells and GC tissues by IF staining, with GC tissues from cohort 1. TCF7L2 protein was detected at varying levels in the cytoplasm and nucleus of both MNK45 cells and GC tissues, whereas PLAUR protein was observed only in the cytoplasm (Figures 5a-b). Through fluorescence intensity, we found that the expression of TCF7L2 and PLAUR were positively correlated in 15 GC tissues (r = 0.54, P < 0.03; Figure 5b). Notably, we observed the co-localization of TCF7L2 and PLAUR proteins in the cytoplasm of GC cells through confocal laser microscopy. Hence, a co-IP assay was employed to further analyze whether there was a protein-protein interaction between TCF7L2 and PLAUR which could regulate the abnormal PLAUR expression in GC. The results revealed an absence of direct interaction between TCF7L2 and PLAUR protein (Supplementary Figure 6). Cell nuclei are considered the main site where transcription events occur in organisms. Overall, it can be deduced that TCF7L2 transcriptionally regulates PLAUR expression.
TCF7L2 promotes transcription of PLAUR in GC. a Representative image of the co-localization of TCF7L2 protein (green) and PLAUR (red) in MKN45 cell lines observed by confocal microscopy. b Representative image of the co-localization of TCF7L2 protein (green) and PLAUR (red) in 15 GC tissues (cohort 1) observed via confocal microscopy; the expression of TCF7L2 and PLAUR was positively correlated via fluorescence quantitative analysis. c-d TCF7L2 has a positive regulatory effect on the expression of PLAUR in GC cells. The effects of TCF7L2 silencing on PLAUR mRNA or protein expression in GC cells as detected by RT-qPCR (c) and western blot (d), respectively. e Motif of the TCF7L2 binding site in the PLAUR promoter, predicted through the JASPAR dataset. f Schematic of the TCF7L2 binding site on the PLAUR promoter. g ChIP-qPCR analysis of TCF7L2 binding to the PLAUR promoter in MKN45 and 293T cell lines. The second position was the most important binding site. e Luciferase activity was determined after mutation of the second TCF7L2 site in the PLAUR promoter in MKN45 and 293T cell lines. The luciferase activity of the wild-type PLAUR promoter was significantly higher than that of mutant PLAUR promoter. Compared with the corresponding control group, * P < 0.001.
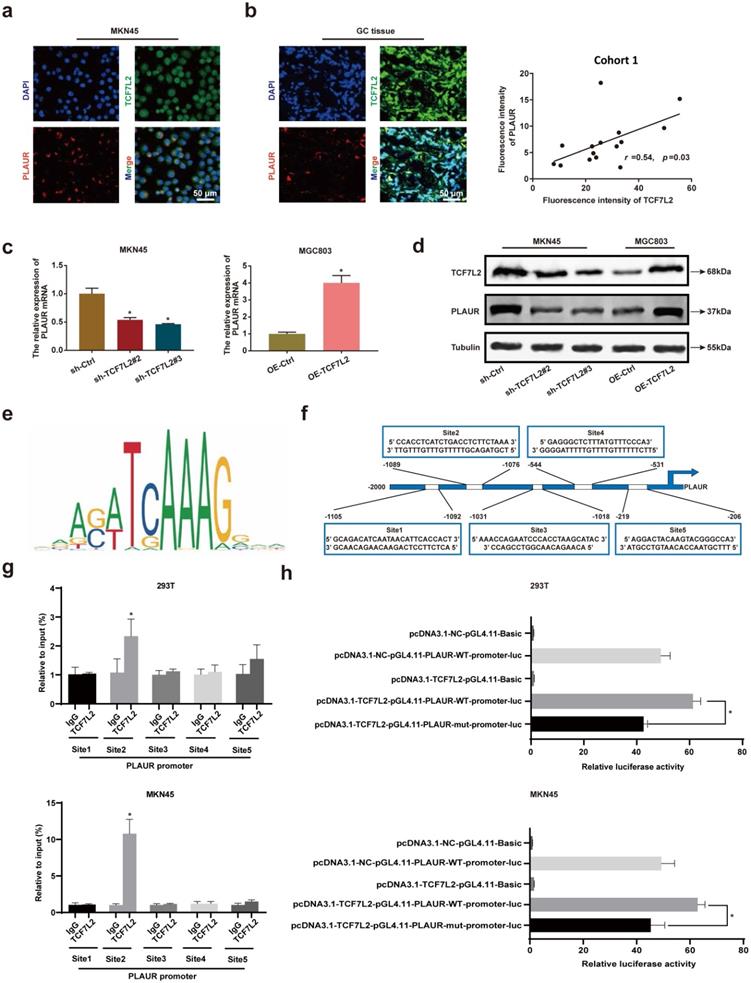
To validate the regulatory effect of TCF7L2 on PLAUR expression, we suppressed TCF7L2 expression in MKN45 cells and overexpressed TCF7L2 in MGC803 cells. The knockdown or overexpression was verified by qRT-PCR and western blot (Supplementary Figure 7). Afterwards, the effects on PLAUR expression were analyzed. The knockdown of TCF7L2 greatly inhibited PLAUR expression in MKN45 cells, whereas its overexpression elevated PLAUR in MGC803 cells (Figure 5c-d). Accordingly, we confirmed that TCF7L2 regulates PLAUR expression in GC.
To assess whether TCF7L2 protein can directly bind to the PLAUR promoter, we identified multiple binding sites using the JASPAR database (Figure 5e; Table S7). For the top 5 binding sites on the JASPAR database (Figure 5f), we performed ChIP analysis in MKN45 and 293T cells. As shown in Figure 5g, the second site (-1089 to -1076) in the PLAUR promoter region was the primary binding site of TCF7L2. Subsequently, to further validate the transcriptional activation effect of TCF7L2 on PLAUR, we again conducted a dual luciferase reporter assay using MKN45 and 293T cells. Based on the ChIP-qPCR results, we mutated the second site in the PLAUR promoter region where TCF7L2 primarily binds and then examined the luciferase activity. The luciferase activity of the wild-type PLAUR promoter was significantly higher than that of the mutant PLAUR promoter (Figure 5h). This suggested that the transcription factor TCF7L2 has a positive regulatory relationship with its downstream target gene, PLAUR. In summary, TCF7L2 promotes PLAUR expression through transcriptional upregulation.
TCF7L2 promotes GC carcinogenesis in vivo and in vitro
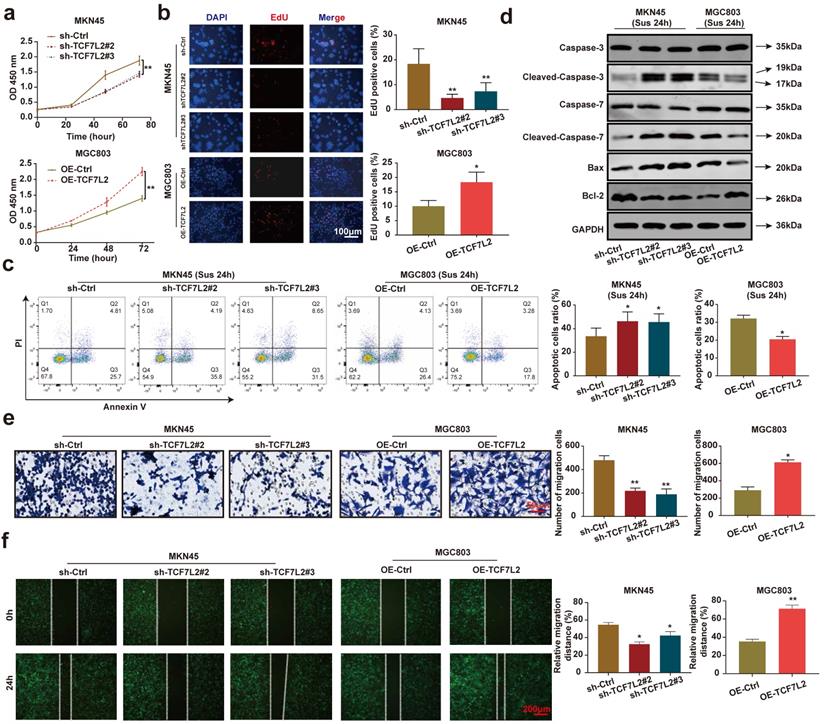
We focused on investigating the effects of TCF7L2 on carcinogenesis in vitro and in vivo. As shown in Figures 6a-b, the knockdown of TCF7L2 inhibited the proliferation of MKN45 cells, whereas its overexpression promoted the proliferation of MGC803 cells. After 24 h suspension culture, the knockdown of TCF7L2 promoted the apoptosis of MKN45 cells, whereas its overexpression inhibited the apoptosis of MGC803 cells (Figure 6c). Western blot results showed that after suspension culture for 24 h, the knockdown of TCF7L2 promoted the expression of cleaved-caspase-3/7 and Bax in MKN45 cells, while inhibiting Bcl-2 expression. In contrast, the overexpression of TCF7L2 inhibited the expression of cleaved-caspase-3/7 and Bax in MGC803 cells, while promoting Bcl-2 expression (Figure 6d). As expected, the knockdown of TCF7L2 inhibited the migration of MKN45 cells, whereas its overexpression promoted the migration of MGC803 cells (Figure 6e-f). Briefly, our data indicated that TCF7L2 affects the proliferation, anoikis resistance, and migration of GC cells.
TCF7L2 promotes the malignant phenotype of GC cells. a-b Effects of TCF7L2 knockdown or overexpression on the proliferation of GC cells measured by CCK-8 (a) and EdU assay (b). c After GC cells were suspended for 24h, the effects of TCF7L2 knockdown or overexpression on the anoikis resistance of GC cells were analyzed via flow cytometry. d Changes in apoptosis-related proteins after TCF7L2 knockdown or overexpression were analyzed via western blot in GC cells suspended for 24 h. e-f Effects of TCF7L2 knockdown or overexpression on the migration of GC cells as evaluated by transwell (e) and wound healing assay (f). Compared with the corresponding control group, * P < 0.05, ** P < 0.001.
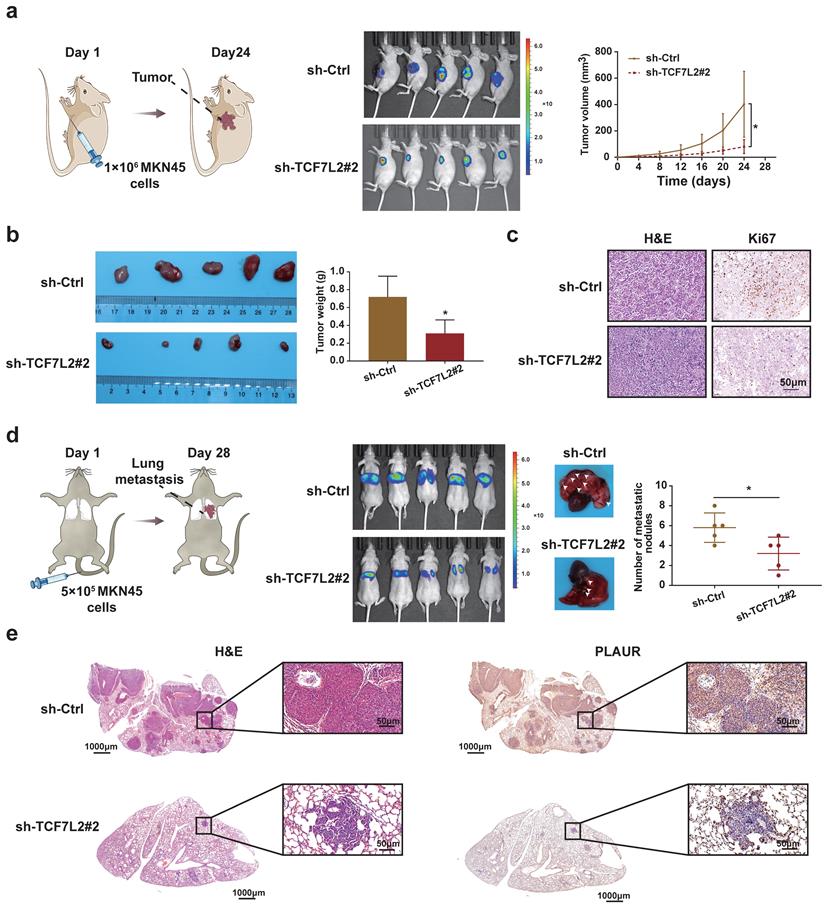
To clarify the functions of TCF7L2 in vivo, we established nude mouse models of subcutaneous tumor and lung metastasis by injecting MKN45 cells, according to previous reports. As shown in Figure 7a, when MKN45 cells with sh-Ctrl or sh-TCF7L2#2 were injected, the fluorescence signal intensity and tumor growth rate of the subcutaneous tumors in the sh-Ctrl group were significantly higher than those in the sh-TCF7L2#2 group. The weight of the tumors was calculated after euthanizing the mice. The sh-Ctrl group exhibited significantly heavier tumors than the sh-TCF7L2#2 group (Figure 7b). We also identified tumors via H&E staining. IHC results indicated weaker tumor tissue expression of Ki67 in the sh-TCF7L2 group than in the sh-Ctrl group (Figure 7c). On the 28th day following tail vein injection of MKN45 cells, the fluorescence signal intensity in the lung from metastatic tumors was significantly higher in the sh-Ctrl group than that in the sh-TCF7L2 group. We also observed fewer average numbers of nodules on the lung surfaces for mice in the sh-TCF7L2 group than those in the sh-Ctrl group (Figure 7d). Importantly, IHC staining revealed that the knockdown of TCF7L2 inhibited PLAUR expression in lung metastasis nodules of the mice (Figure 7e). In summary, TCF7L2 promoted the growth and metastasis of GC as well as regulated the expression of PLAUR in vivo.
TCF7L2 promotes the GC process in vivo. a Knockdown of TCF7L2 inhibited the growth of subcutaneous xenograft tumors in nude mice. Bioluminescence images of the nude mice subcutaneous tumors were displayed through a small animal in vivo imaging system, and the tumor growth curve was measured for 24 days. b Gross specimen and average weight of the tumor after animals were sacrificed. Knockdown of TCF7L2 inhibited the volume and weight of subcutaneous xenograft tumors in nude mice. c Histological characteristics and Ki67 expression of the tumor were examined via H&E staining and IHC, respectively. Knockdown of TCF7L2 inhibited the Ki67 expression in subcutaneous xenograft tumors. d Knockdown of TCF7L2 inhibited lung metastasis. The bioluminescence image of lung metastases and the count of lung metastatic nodules was obtained 28 days after tail veins were injected with GC cells. The white arrows represent the lung metastasis nodule. e Lung metastases were confirmed via H&E staining (left); knockdown of TCF7L2 inhibited PLAUR expression in lung metastases evaluated by IHC (right). Compared with the control group, * P < 0.001.
Role of PLAUR in TCF7L2 induction of GC proliferation, anoikis resistance, and migration
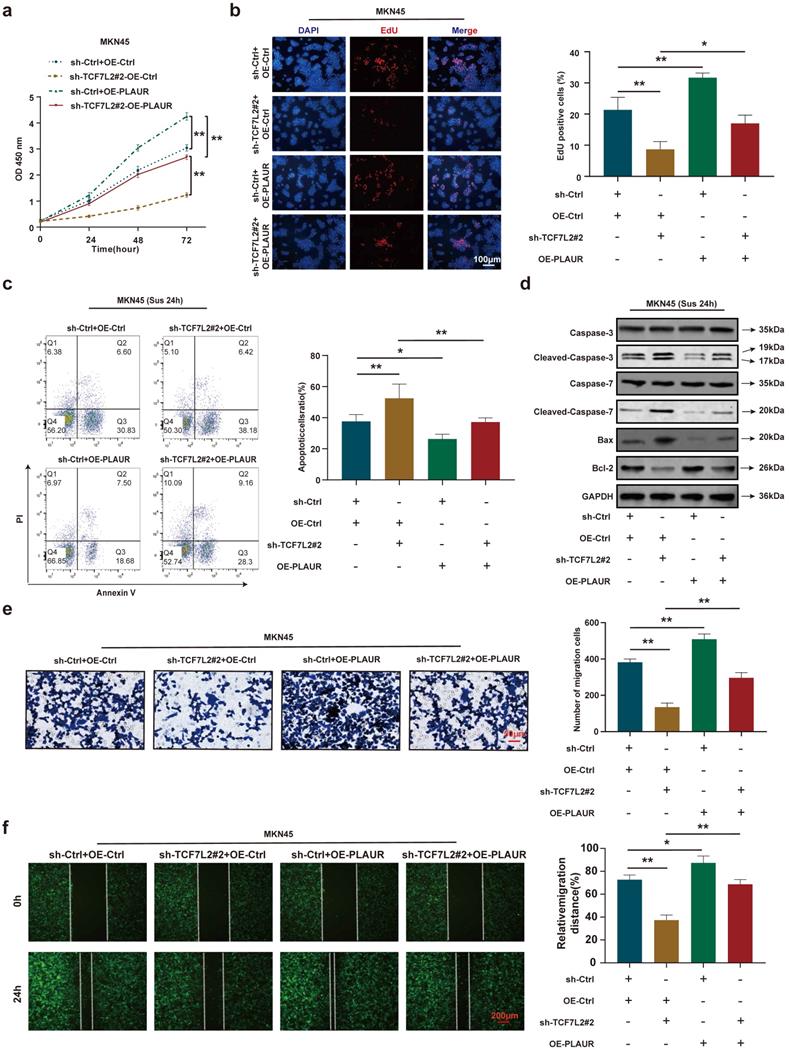
To further validate that PLAUR is a downstream target gene of TCF7L2, we again performed rescue experiments using sh-TCF7L2 MKN45 cells to overexpress PLAUR and observed the changes in proliferation, anoikis resistance, and cell migration. CCK8 and EdU assays demonstrated that TCF7L2 knockdown inhibited the proliferation of MKN45 cells, and the reduction of MKN45 cell proliferation by sh-TCF7L2 was reversed by co-transfection with PLAUR (Figure 8a-b). As shown in Figure 8c, the knockdown of TCF7L2 inhibited the anoikis resistance of MKN45 cells, which was partially reversed by co-transfection with PLAUR. Western blot revealed that when MKN45 cells were suspended for 24 h, the sh-TCF7L2-induced increases in cleaved-caspase-3/7 and Bax expression were partially reversed by the overexpression of PLAUR, and the sh-TCF7L2-induced decrease in Bcl-2 expression was also partially reversed by PLAUR overexpression (Figure 8d). In transwell and wound healing assays, TCF7L2 knockdown inhibited the migration capacity of MKN45 cells, and the sh-TCF7L2-induced reduction of this migration capacity was partially rescued by co-transfection with PLAUR (Figure 8e-f). These results indicated that TCF7L2 stimulates the proliferation, anoikis resistance, and migration of GC cells in a PLAUR-dependent manner.
PLAUR is essential for the malignant phenotype of GC cells mediated by TCF7L2. a-b Cell proliferation of MKN45 cells with knockdown of TCF7L2 or control, and re-expression of PLAUR in TCF7L2 depleted cells, as quantified by CCK-8 (a) and EdU (b) assay, respectively. c After GC cells were suspended for 24 h, the anoikis resistance of MKN45 cells with knockdown of TCF7L2 or control and re-expression of PLAUR in TCF7L2 depleted cells were assessed via flow cytometry. d After GC cells were suspended for 24 h, western blot for apoptosis-related proteins in MKN45 cells with the depletion of TCF7L2 or control, or re-expression of PLAUR in TCF7L2 depleted cells. e-f Measurement of cell migration by transwell (e) and wound healing (f) assays using MKN45 cells with depletion of TCF7L2 or control, or re-expression of PLAUR in TCF7L2 depleted cells. * P<0.05, ** P<0.001.
TCF7L2 is associated with poor prognosis of patients with GC
Finally, this study assessed the effects of TCF7L2 on the prognosis of patients with GC. TCF7L2 protein expression in 121 pairs of GC and adjacent tissues (cohort 2) was examined via IHC. As shown in Figure 9a, TCF7L2 protein was stained to varying degrees in both the cell cytoplasm and nucleus. Notably, TCF7L2 protein was primarily localized in the cell nuclei of GC tissue, as well as in the cytoplasm in adjacent tissues. This suggested that TCF7L2 exerts a cancer-promoting role in the nucleus of GC cells. A high expression of TCF7L2 was observed in 46.3% (56/121) of GC tissues, while showing a significant difference (χ2 = 6.305, P = 0.012) in only 30.6% (37/121) of adjacent tissues. Meanwhile, we quantitatively explored the expression levels of TCF7L2 protein in 10 pairs of fresh GC and adjacent tissues (cohort 3) via western blot. As shown in Figure 9b, TCF7L2 protein was more highly expressed in GC tissues than in adjacent tissues (P < 0.001). Consistent with prior bioinformatics results, TCF7L2 appeared to be upregulated in GC tissues.
High TCF7L2 expression predicts poor prognosis of patients with GC. a Typical images of TCF7L2 protein in GC and adjacent tissues stained by IHC. In GC tissues, TCF7L2 protein was located in the nucleus and cytoplasm; in adjacent tissues, TCF7L2 protein was primarily located in cytoplasm. b Survival curve of 121 GC patients (cohort 2). High TCF7L2 expression correlated with poor prognosis of GC patients. c TCF7L2 protein was quantitatively analyzed via western blot in 10 pairs of fresh GC and adjacent tissues (cohort 3). d Independent prognostic factor for overall survival of 121 patients with GC, shown as a forest plot.
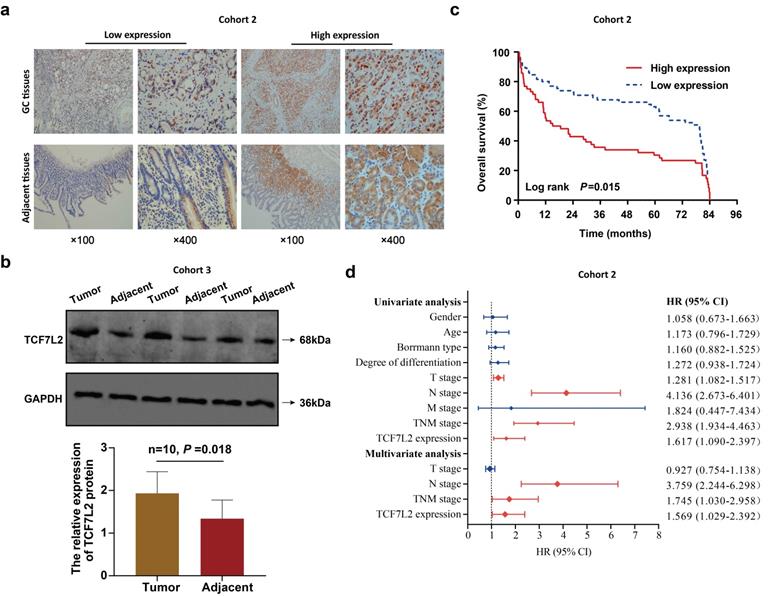
For the analysis of the correlation between TCF7L2 and patient prognosis, we divided the 121 patients with GC into TCF7L2 “high expression” (n = 56) and “low expression” (n = 65) groups. Meanwhile, we collected and statistically analyzed the clinicopathological data of all patients with GC and found that high TCF7L2 expression was closely linked to lymph node metastasis and TNM staging (P < 0.05); however, it was not linked to such clinicopathological features such as gender, age, tumor size, Borrmann classification, tumor location, and degree of differentiation (P > 0.05) (Table 1). Follow-up analyses revealed that the survival time of patients with GC with low TCF7L2 expression was markedly longer than those with high TCF7L2 expression (Figure 9c). Finally, we established a Cox regression model by utilizing the clinicopathological and survival data of 121 patients with GC to assess whether TCF7L2 could act as an independent risk predictor for the prognosis of patients with GC. Univariate analysis revealed that deeper tumor invasion (T stage), lymph node metastasis (N stage), advanced TNM stage, and high TCF7L2 expression were significantly correlated with a poor prognosis for patients with GC (Figure 9d). According to the multivariate analysis, high TCF7L2 expression could serve as an independent risk predictor for poor prognosis of patients with GC (P = 0.044).
The relationship between TCF7L2 expression and clinicopathological characteristics of patients with GC
| Factor | TCF7L2 expression | χ2 | P-value | |
|---|---|---|---|---|
| Low (n=65) | High (n=56) | |||
| Gender | 2.690 | 0.101 | ||
| Male | 45 (69.2) | 46 (82.1) | ||
| Female | 20 (30.8) | 10 (17.9) | ||
| Age | 0.132 | 0.716 | ||
| ≤60y | 35 (53.8) | 32 (57.1) | ||
| >60y | 30 (46.2) | 24 (42.9) | ||
| Tumor size | 0.963 | 0.326 | ||
| ≤5cm | 45 (69.2) | 34 (60.7) | ||
| >5cm | 20 (30.8) | 22 (39.3) | ||
| Borrmann type | 1.413 | 0.703 | ||
| I | 9 (13.8) | 4 (7.1) | ||
| II | 13 (20) | 12 (21.4) | ||
| III | 42 (64.6) | 39 (69.6) | ||
| IV | 1 (1.5) | 1 (1.8) | ||
| Type of gastrectomy | 2.354 | 0.671 | ||
| Gastrectomy-partial | 10 (15.4) | 10 (17.9) | ||
| Near-total gastrectomy | 34 (52.3) | 22 (39.3) | ||
| Total gastrectomy | 18 (27.7) | 20 (35.7) | ||
| Gastrectomy with esophagus | 1 (1.5) | 2 (3.6) | ||
| Gastrectomy with other organs | 2 (3.1) | 2 (3.6) | ||
| Degree of differentiation | 1.644 | 0.439 | ||
| Well | 3 (4.6) | 6 (10.7) | ||
| Moderate | 24 (36.9) | 20 (35.7) | ||
| Poor | 38 (58.5) | 30 (53.6) | ||
| T stage | 2.478 | 0.649 | ||
| Tis | 1 (1.5) | 3 (5.4) | ||
| T1 | 6 (9.2) | 7 (12.5) | ||
| T2 | 9 (13.8) | 7 (12.5) | ||
| T3 | 8 (12.3) | 4 (7.1) | ||
| T4 | 41 (63.1) | 35 (62.5) | ||
| N stage | 4.447 | 0.035 | ||
| N0 | 32 (49.2) | 17 (30.4) | ||
| N1-N3 | 33 (50.8) | 39 (69.6) | ||
| M stage | 0.011 | 0.915 | ||
| M0 | 64 (98.5) | 55 (98.2) | ||
| M1 | 1 (1.5) | 1 (1.8) | ||
| TNM stage | 4.682 | 0.030 | ||
| I-II | 36 (55.4) | 20 (35.7) | ||
| III-IV | 29 (44.6) | 36 (64.3) | ||
Discussion
This study revealed that TCF7L2 was highly expressed in GC, which is closely associated with the poor clinicopathological features of patients. High TCF7L2 expression is an independent risk factor for poor prognosis in patients with GC. In vitro, we confirmed that TCF7L2 promotes the proliferation, anoikis resistance, and migration of GC cells. In vivo, we found that TCF7L2 promoted tumor growth and metastasis in nude mice. Importantly, TCF7L2 was found to be a major transcriptional regulator of PLAUR, with binding sites within the promoter region of PLAUR, leading to its transcriptional activation. Similarly, we also verified that PLAUR promotes the malignant behaviors of GC in vitro and in vivo, particularly the anoikis resistance of GC cells. Altogether, our data revealed that the transcription factor TCF7L2 is overexpressed and abnormally activates PLAUR in GC and is involved in promoting the anoikis resistance and migration of GC cells.
Members of the fibrinolytic system play important roles in cellular chemotaxis, invasion, migration, regulation of various cytokines and growth factors, immune response, and angiogenesis [13, 14, 43, 44]. PLAUR, as one of the major members of this family, also plays important roles in the malignant biological process of GC, particularly the migration of GC cells. It has been established that the collagen fibrin network is formed in the intercellular space via stromal cell secretion, which effectively prevents the migration of tumor cells, owing to its intricate structure [45, 46]. Tumor cells destroy the aforementioned stromal network by activating the fibrinolytic system through PLAUR, thereby creating favorable conditions for metastasis. Through semi-quantitative IHC, Heiss et al. [47] revealed that PLAUR expression was increased in GC tissues. Based on Cox analysis, they further found that PLAUR was significantly negatively correlated with the survival time of patients with GC. Additionally, many other studies have confirmed that PLAUR is highly expressed in GC, exhibiting a close association with the distant metastasis of tumors [48-50]. Thus, PLAUR is an important regulator of GC cell migration. In this study, we verified that PLAUR promotes the malignant process of GC in both in vitro and in vivo experiments. Notably, to the best of our knowledge, there have been no previous reports on the correlation between PLAUR and anoikis resistance of GC cells. Nevertheless, in cases like melanoma and prostatic cancer, researchers have proposed a relationship between PLAUR and anoikis resistance [51-53]. We used an anoikis model with GC cells suspended for 24 h and further validated the ability of PLAUR to promote the anoikis resistance of GC cells.
Furthermore, this study explored the molecular mechanism whereby PLAUR is upregulated in GC. We analyzed the reasons for the high PLAUR expression from an abnormal transcription perspective, identifying TCF7L2 as a potential transcriptional regulator of PLAUR. We found that TCF7L2 protein was localized primarily in the nuclei of GC cells. Thereafter, through ChIP-qPCR and dual luciferase reporter assays, we not only confirmed the presence of binding sites between TCF7L2 and PLAUR promoter region, but also verified that TCF7L2 positively regulated PLAUR expression in GC via transcription. PLAUR is also regulated by other transcription factors. For example, FOXM1c is upregulated in pancreatic cancer which, as a transcription factor, has binding sites within the PLAUR promoter and causes the upregulation of PLAUR, ultimately affecting the migration of pancreatic cancer cells [57]. Based on luciferase assays, researchers found that the hypoxia-induced overexpression of HIF-1α activated the transcriptional activity of the PLAUR promoter, thereby enhancing its expression in cervical cancer, so that the cervical cancer cells were more invasive under hypoxic conditions [58]. In hepatoma cells, ETV4 directly binds to the core region of the PLAUR promoter, while PBK can enhance the binding between ETV4 and the PLAUR promoter [59]. However, the role of TCF7L2 in GC remained unclear, particularly its correlation with the anoikis resistance of GC cells.
TCF7L2, as a downstream effector of Wnt/β-catenin signaling pathway, contains a highly conserved HMG box and a small basic peptide motif, similar to other members of its family [60, 61]. Together, these two structures constitute the HMG DNA domain, which can recognize specific DNA sequences [60, 62, 63]. Additionally, the members of this family also contain a β-catenin binding domain and a C-clamp domain [60, 64, 65]. The C-clamp domain is located at the carboxyl terminal of the HMG DNA domain and has a specific DNA binding activity [60, 66]. Thus, TCF7L2 has two specific DNA binding domains. In various solid tumors, TCF7L2 is abnormally expressed and is involved in the malignant process. Compared to low-grade gliomas, TCF7L2 is upregulated in glioblastoma tissues, exhibiting a close association with the poor prognosis of patients with glioblastoma [67]. In pancreatic cancer, high TCF7L2 expression predicts a poor prognosis. TCF7L2 leads to the upregulation of HIF-1α by inhibiting the expression of EGLN2, an Egl-9 family member, thereby positively regulating aerobic glycolysis [68]. As a transcriptional activator of WNT7B, TCF7L2 binds to its promoter region to further facilitate the growth and migration of GC cells [69]. The long non-coding RNA SNHG11 can enhance the oncogenic autophagy of GC cells, thereby promoting the proliferation, stemness, migration, invasion, and epithelial-mesenchymal transition of tumor cells. Its upregulation in GC is achieved by transcriptional activation of TCF7L2 [70]. Our study revealed that TCF7L2 is upregulated in GC tissues and cells. Through in vitro cell experimentation, we verified that TCF7L2 promotes the proliferation, anoikis resistance, and migration capacity of GC cells. In our in vivo experiments, TCF7L2 promoted tumor growth and metastasis in nude mice. Notably, anoikis resistance is a key pre-step for distant tumor metastasis, and TCF7L2 was closely associated with the anoikis resistance of GC cells. Through functional rescue experiments, this study also confirmed that TCF7L2 plays an important role in the malignant process of GC in a PLAUR-dependent manner.
Conclusions
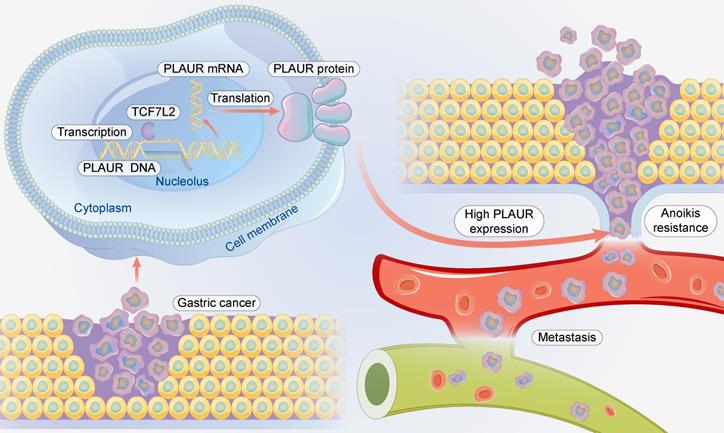
In conclusion, TCF7L2 is highly expressed in GC and is an independent risk predictor for the poor prognosis of patients with GC. TCF7L2 enhances the proliferation, anoikis resistance, and migration of GC cells in a PLAUR-dependent manner (Figure 10). Our study clarifies that PLAUR affects the upstream molecular regulation of GC, which also provides a theoretical basis for the use of TCF7L2 and PLAUR as potential therapeutic targets for GC.
Schematic summary. Mechanistically, TCF7L2 is upregulated in GC and transcriptionally activates PLAUR, which promotes anoikis resistance and metastasis of GC cells.
Abbreviations
ChIP: Chromatin-immunoprecipitation; co-IP: Co-immunoprecipitation; DAPI: 4',6-diamidino-2-phenylindole; EdU: 5-ethynyl-2'-deoxyuridine; GC: gastric cancer; IHC: immunohistochemistry; PLAUR: urokinase-type plasminogen activator receptor; TCF7L2: transcription Factor 7 Like 2; HMG: high-mobility group; FBS: fetal bovine serum; shRNA: small hairpin RNA; qRTPCR: quantitative reverse transcription polymerase chain reaction; PVDF: polyvinylidene difluoride; IF: immunofluorescence; LC-MS/MS: liquid chromatography tandem mass spectrometry; H&E: hematoxylin and eosin; IHC: immunohistochemistry; RPMI: Roswell Park Memorial Institute; RIPA: radio immunoprecipitation assay; STAD: stomach adenocarcinoma.
Supplementary Material
Supplementary figures and tables 1, 2, 3, 4, 5, and 7.
Supplementary table 6.
Acknowledgements
We acknowledge TCGA, UCSC, PROMO, JASPAR, GEPIA, HPA, and Kaplan-Meier plotter database for providing their platforms and contributors for uploading their meaningful datasets.
Funding
Supported by grants from (1) National Natural Science Foundation of China (81670594; 81470791; 81376597; 81802269); (2) Major Science and Technology Project of Gansu Province (19ZD2WA001); (3) Natural Science Foundation of Gansu Province (1606RJIA328; 20JR5RA352; 20JR10RA686; 21JR7RA386); (4) Gansu Province Higher Education Innovation Ability Improvement Project (2019B-009; 2020B-009).
Author contributions
TZ and HC conceived and supervised the project. TZ, BW and HC designed the experiments of this study. TZ, BW, BG, LX and XL performed the experiments. TZ, FS, LG, PZ and XL contributed to the data analysis. BW, LG, and LX collected the clinical data and tissue samples. TZ and LG maintained the mice. TZ, BW and HC participated in the revision of the manuscript. TZ, FS and BG wrote and edited of the manuscript and approved the final draft of the manuscript.
Availability of data and materials
The data in the current study is available from the corresponding author upon reasonable requests.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: a cancer journal for clinicians. 2021;71:209-49
2. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F. et al. Cancer statistics in China, 2015. CA: a cancer journal for clinicians. 2016;66:115-32
3. Steeg PS. Targeting metastasis. Nature reviews Cancer. 2016;16:201-18
4. Kim YN, Koo KH, Sung JY, Yun UJ, Kim H. Anoikis resistance: an essential prerequisite for tumor metastasis. International journal of cell biology. 2012;2012:306879
5. Simpson CD, Anyiwe K, Schimmer AD. Anoikis resistance and tumor metastasis. Cancer letters. 2008;272:177-85
6. Taddei ML, Giannoni E, Fiaschi T, Chiarugi P. Anoikis: an emerging hallmark in health and diseases. The Journal of pathology. 2012;226:380-93
7. Cardone MH, Salvesen GS, Widmann C, Johnson G, Frisch SM. The regulation of anoikis: MEKK-1 activation requires cleavage by caspases. Cell. 1997;90:315-23
8. Zhang HF, Hughes CS, Li W, He JZ, Surdez D, El-Naggar AM. et al. Proteomic Screens for Suppressors of Anoikis Identify IL1RAP as a Promising Surface Target in Ewing Sarcoma. Cancer discovery. 2021;11:2884-903
9. Ye G, Yang Q, Lei X, Zhu X, Li F, He J. et al. Nuclear MYH9-induced CTNNB1 transcription, targeted by staurosporin, promotes gastric cancer cell anoikis resistance and metastasis. Theranostics. 2020;10:7545-60
10. Ichikawa T, Okugawa Y, Toiyama Y, Tanaka K, Yin C, Kitajima T. et al. Clinical significance and biological role of L1 cell adhesion molecule in gastric cancer. British journal of cancer. 2019;121:1058-68
11. Falanga A, Marchetti M, Vignoli A. Coagulation and cancer: biological and clinical aspects. Journal of thrombosis and haemostasis: JTH. 2013;11:223-33
12. Li Santi A, Napolitano F, Montuori N, Ragno P. The Urokinase Receptor: A Multifunctional Receptor in Cancer Cell Biology. Therapeutic Implications. International journal of molecular sciences. 2021 22
13. Mahmood N, Rabbani SA. Fibrinolytic System and Cancer: Diagnostic and Therapeutic Applications. International journal of molecular sciences. 2021 22
14. Choi JW, Kim JK, Yang YJ, Kim P, Yoon KH, Yun SH. Urokinase exerts antimetastatic effects by dissociating clusters of circulating tumor cells. Cancer research. 2015;75:4474-82
15. Kwaan HC, Lindholm PF. Fibrin and Fibrinolysis in Cancer. Seminars in thrombosis and hemostasis. 2019;45:413-22
16. Alpízar-Alpízar W, Nielsen BS, Sierra R, Illemann M, Ramírez JA, Arias A. et al. Urokinase plasminogen activator receptor is expressed in invasive cells in gastric carcinomas from high- and low-risk countries. International journal of cancer. 2010;126:405-15
17. Beyer BC, Heiss MM, Simon EH, Gruetzner KU, Babic R, Jauch KW. et al. Urokinase system expression in gastric carcinoma: prognostic impact in an independent patient series and first evidence of predictive value in preoperative biopsy and intestinal metaplasia specimens. Cancer. 2006;106:1026-35
18. Tincknell G, Piper AK, Aghmesheh M, Becker T, Vine KL, Brungs D. et al. Experimental and Clinical Evidence Supports the Use of Urokinase Plasminogen Activation System Components as Clinically Relevant Biomarkers in Gastroesophageal Adenocarcinoma. Cancers. 2021 13
19. Dias C, Pfundt R, Kleefstra T, Shuurs-Hoeijmakers J, Boon EMJ, van Hagen JM. et al. De novo variants in TCF7L2 are associated with a syndromic neurodevelopmental disorder. American journal of medical genetics Part A. 2021;185:2384-90
20. Nguyen-Tu MS, Martinez-Sanchez A, Leclerc I, Rutter GA, da Silva Xavier G. Adipocyte-specific deletion of Tcf7l2 induces dysregulated lipid metabolism and impairs glucose tolerance in mice. Diabetologia. 2021;64:129-41
21. Savic D, Ye H, Aneas I, Park SY, Bell GI, Nobrega MA. Alterations in TCF7L2 expression define its role as a key regulator of glucose metabolism. Genome research. 2011;21:1417-25
22. Zhou Y, Park SY, Su J, Bailey K, Ottosson-Laakso E, Shcherbina L. et al. TCF7L2 is a master regulator of insulin production and processing. Human molecular genetics. 2014;23:6419-31
23. Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R. et al. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell. 2008;135:37-48
24. Zhang Z, Xu L, Xu X. The role of transcription factor 7-like 2 in metabolic disorders. Obesity reviews: an official journal of the International Association for the Study of Obesity. 2021;22:e13166
25. da Silva Xavier G, Mondragon A, Mourougavelou V, Cruciani-Guglielmacci C, Denom J, Herrera PL. et al. Pancreatic alpha cell-selective deletion of Tcf7l2 impairs glucagon secretion and counter-regulatory responses to hypoglycaemia in mice. Diabetologia. 2017;60:1043-50
26. McCarthy MI, Rorsman P, Gloyn AL. TCF7L2 and diabetes: a tale of two tissues, and of two species. Cell metabolism. 2013;17:157-9
27. Chang J, Tian J, Yang Y, Zhong R, Li J, Zhai K. et al. A Rare Missense Variant in TCF7L2 Associates with Colorectal Cancer Risk by Interacting with a GWAS-Identified Regulatory Variant in the MYC Enhancer. Cancer research. 2018;78:5164-72
28. Chen J, Yuan T, Liu M, Chen P. Association between TCF7L2 gene polymorphism and cancer risk: a meta-analysis. PloS one. 2013;8:e71730
29. Drake I, Wallström P, Hindy G, Ericson U, Gullberg B, Bjartell A. et al. TCF7L2 type 2 diabetes risk variant, lifestyle factors, and incidence of prostate cancer. The Prostate. 2014;74:1161-70
30. Slattery ML, Folsom AR, Wolff R, Herrick J, Caan BJ, Potter JD. Transcription factor 7-like 2 polymorphism and colon cancer. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2008;17:978-82
31. Wang Y, Men X, Gu Y, Wang H, Xu Z. Haplotype analysis on correlation between transcription factor 7-like 2 gene polymorphism and breast cancer risk. BMC cancer. 2021;21:885
32. Huang T, Chen B, Wang F, Cai W, Wang X, Huang B. et al. Rab1A promotes IL-4R/JAK1/STAT6-dependent metastasis and determines JAK1 inhibitor sensitivity in non-small cell lung cancer. Cancer letters. 2021;523:182-94
33. Chen P, Tao L, Wang T, Zhang J, He A, Lam KH. et al. Structural basis for recognition of frizzled proteins by Clostridium difficile toxin B. Science (New York, NY). 2018;360:664-9
34. Duan Y, Huo D, Gao J, Wu H, Ye Z, Liu Z. et al. Ubiquitin ligase RNF20/40 facilitates spindle assembly and promotes breast carcinogenesis through stabilizing motor protein Eg5. Nature communications. 2016;7:12648
35. Gao X, Jiang M, Chu Y, Han Y, Jin Y, Zhang W. et al. ETV4 promotes pancreatic ductal adenocarcinoma metastasis through activation of the CXCL13/CXCR5 signaling axis. Cancer letters. 2022;524:42-56
36. Skidmore ZL, Wagner AH, Lesurf R, Campbell KM, Kunisaki J, Griffith OL. et al. GenVisR: Genomic Visualizations in R. Bioinformatics (Oxford, England). 2016;32:3012-4
37. Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D. et al. The UCSC Table Browser data retrieval tool. Nucleic acids research. 2004;32:D493-6
38. Messeguer X, Escudero R, Farré D, Núñez O, Martínez J, Albà MM. PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics (Oxford, England). 2002;18:333-4
39. Castro-Mondragon JA, Riudavets-Puig R, Rauluseviciute I, Berhanu Lemma R, Turchi L, Blanc-Mathieu R. et al. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic acids research. 2021
40. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic acids research. 2017;45:W98-w102
41. Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A. et al. Proteomics. Tissue-based map of the human proteome. Science (New York, NY). 2015;347:1260419
42. Szász AM, Lánczky A, Nagy Á, Förster S, Hark K, Green JE. et al. Cross-validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1,065 patients. Oncotarget. 2016;7:49322-33
43. Heissig B, Eiamboonsert S, Salama Y, Shimazu H, Dhahri D, Munakata S. et al. Cancer therapy targeting the fibrinolytic system. Advanced drug delivery reviews. 2016;99:172-9
44. Mondino A, Blasi F. uPA and uPAR in fibrinolysis, immunity and pathology. Trends in immunology. 2004;25:450-5
45. Heissig B, Dhahri D, Eiamboonsert S, Salama Y, Shimazu H, Munakata S. et al. Role of mesenchymal stem cell-derived fibrinolytic factor in tissue regeneration and cancer progression. Cellular and molecular life sciences: CMLS. 2015;72:4759-70
46. Tian C, Huang Y, Clauser KR, Rickelt S, Lau AN, Carr SA. et al. Suppression of pancreatic ductal adenocarcinoma growth and metastasis by fibrillar collagens produced selectively by tumor cells. Nature communications. 2021;12:2328
47. Heiss MM, Babic R, Allgayer H, Gruetzner KU, Jauch KW, Loehrs U. et al. Tumor-associated proteolysis and prognosis: new functional risk factors in gastric cancer defined by the urokinase-type plasminogen activator system. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 1995;13:2084-93
48. Kaneko T, Konno H, Baba M, Tanaka T, Nakamura S. Urokinase-type plasminogen activator expression correlates with tumor angiogenesis and poor outcome in gastric cancer. Cancer science. 2003;94:43-9
49. Park IK, Kim BJ, Goh YJ, Lyu MA, Park CG, Hwang ES. et al. Co-expression of urokinase-type plasminogen activator and its receptor in human gastric-cancer cell lines correlates with their invasiveness and tumorigenicity. International journal of cancer. 1997;71:867-73
50. Plebani M, Herszènyi L, Carraro P, De Paoli M, Roveroni G, Cardin R. et al. Urokinase-type plasminogen activator receptor in gastric cancer: tissue expression and prognostic role. Clinical & experimental metastasis. 1997;15:418-25
51. Alfano D, Iaccarino I, Stoppelli MP. Urokinase signaling through its receptor protects against anoikis by increasing BCL-xL expression levels. The Journal of biological chemistry. 2006;281:17758-67
52. Hasanuzzaman M, Kutner R, Agha-Mohammadi S, Reiser J, Sehgal I. A doxycycline-inducible urokinase receptor (uPAR) upregulates uPAR activities including resistance to anoikis in human prostate cancer cell lines. Molecular cancer. 2007;6:34
53. Sehgal I, Foster TP, Francis J. Prostate cancer cells show elevated urokinase receptor in a mouse model of metastasis. Cancer cell international. 2006;6:21
54. Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell. 2013;152:1237-1251
55. Bradner JE, Hnisz D, Young RA. Transcriptional Addiction in Cancer. Cell. 2017: 168; 629-643.
56. Littlewood TD, Kreuzaler P, Evan GI. All things to all people. Cell. 2012: 151; 11-13.
57. Huang C, Xie D, Cui J, Li Q, Gao Y, Xie K. FOXM1c promotes pancreatic cancer epithelial-to-mesenchymal transition and metastasis via upregulation of expression of the urokinase plasminogen activator system. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:1477-88
58. Nishi H, Sasaki T, Nagamitsu Y, Terauchi F, Nagai T, Nagao T. et al. Hypoxia inducible factor-1 mediates upregulation of urokinase-type plasminogen activator receptor gene transcription during hypoxia in cervical cancer cells. Oncology reports. 2016;35:992-8
59. Yang QX, Zhong S, He L, Jia XJ, Tang H, Cheng ST. et al. PBK overexpression promotes metastasis of hepatocellular carcinoma via activating ETV4-uPAR signaling pathway. Cancer letters. 2019;452:90-102
60. Cadigan KM, Waterman ML. TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harbor perspectives in biology. 2012 4
61. Love JJ, Li X, Case DA, Giese K, Grosschedl R, Wright PE. Structural basis for DNA bending by the architectural transcription factor LEF-1. Nature. 1995;376:791-5
62. Atcha FA, Syed A, Wu B, Hoverter NP, Yokoyama NN, Ting JH. et al. A unique DNA binding domain converts T-cell factors into strong Wnt effectors. Molecular and cellular biology. 2007;27:8352-63
63. van Beest M, Dooijes D, van De Wetering M, Kjaerulff S, Bonvin A, Nielsen O. et al. Sequence-specific high mobility group box factors recognize 10-12-base pair minor groove motifs. The Journal of biological chemistry. 2000;275:27266-73
64. Hecht A, Stemmler MP. Identification of a promoter-specific transcriptional activation domain at the C terminus of the Wnt effector protein T-cell factor 4. The Journal of biological chemistry. 2003;278:3776-85
65. Wenzel J, Rose K, Haghighi EB, Lamprecht C, Rauen G, Freihen V. et al. Loss of the nuclear Wnt pathway effector TCF7L2 promotes migration and invasion of human colorectal cancer cells. Oncogene. 2020;39:3893-909
66. Weise A, Bruser K, Elfert S, Wallmen B, Wittel Y, Wöhrle S. et al. Alternative splicing of Tcf7l2 transcripts generates protein variants with differential promoter-binding and transcriptional activation properties at Wnt/beta-catenin targets. Nucleic acids research. 2010;38:1964-81
67. Jing S, Chen L, Han S, Liu N, Han M, Yang Y. et al. Expression of TCF7L2 in Glioma and Its Relationship With Clinicopathological Characteristics and Patient Overall Survival. Frontiers in neurology. 2021;12:627431
68. Xiang J, Hu Q, Qin Y, Ji S, Xu W, Liu W. et al. TCF7L2 positively regulates aerobic glycolysis via the EGLN2/HIF-1α axis and indicates prognosis in pancreatic cancer. Cell death & disease. 2018;9:321
69. Gao Q, Yang L, Shen A, Li Y, Li Y, Hu S. et al. A WNT7B-m(6)A-TCF7L2 positive feedback loop promotes gastric cancer progression and metastasis. Signal transduction and targeted therapy. 2021;6:43
70. Wu Q, Ma J, Wei J, Meng W, Wang Y, Shi M. lncRNA SNHG11 Promotes Gastric Cancer Progression by Activating the Wnt/β-Catenin Pathway and Oncogenic Autophagy. Molecular therapy: the journal of the American Society of Gene Therapy. 2021;29:1258-78
Author contact
![]() Corresponding author: Cancer center, Lanzhou University Second Hospital, No. 82, Cuiyingmen, Chengguan District, Lanzhou, Gansu Province, 730030, China. E-mail: ery_chenhedu.cn (H. Chen).
Corresponding author: Cancer center, Lanzhou University Second Hospital, No. 82, Cuiyingmen, Chengguan District, Lanzhou, Gansu Province, 730030, China. E-mail: ery_chenhedu.cn (H. Chen).