Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2026; 22(10):5475-5494. doi:10.7150/ijbs.127110 This issue Cite
Research Paper
SQSTM1/p62 UFMylation Enhances Autophagic Clearance of Pathogenic Mutant Huntingtin
Xiaohui Wang1, Xiaowei Lv1, Honglv Jiang1, Wenyun Zhu1, Lindong Cao1, Liang Zhou1, Fang Lin1, Rong Deng2, Li-Fang Hu3, Jingjing Ma4,5 ![]() , Jia-Bin Li1
, Jia-Bin Li1 ![]() , Guoqiang Xu1,6,7,8,9
, Guoqiang Xu1,6,7,8,9 ![]()
1. Jiangsu Key Laboratory of Drug Discovery and Translational Research for Brain Diseases and College of Pharmaceutical Sciences, The Fourth Affiliated Hospital of Soochow University, Jiangsu Province Engineering Research Center of Precision Diagnostics and Therapeutics Development, Jiangsu Key Laboratory of Preventive and Translational Medicine for Major Chronic Non-communicable Diseases, Suzhou Key Laboratory of Drug Research for Prevention and Treatment of Hyperlipidemic Diseases, Soochow University, Suzhou, Jiangsu 215123, China.
2. Department of Neurology and Clinical Research Center of Neurological Disease, The Second Affiliated Hospital of Soochow University, Suzhou, Jiangsu 215004, China.
3. Jiangsu Key Laboratory of Drug Discovery and Translational Research for Brain Diseases, Institute of Neuroscience, Soochow University, Suzhou, Jiangsu 215123, China.
4. Department of Pharmacy, The Fourth Affiliated Hospital of Soochow University, Suzhou Dushu Lake Hospital, Medical Center of Soochow University, Suzhou, Jiangsu 215123, China.
5. State Key Laboratory of Neurology and Oncology Drug Development, Jiangsu, China.
6. Biomedical Basic Research Center (BBRC) of Jiangsu Province, Medical College of Soochow University, Suzhou, Jiangsu 215123, China.
7. Suzhou International Joint Laboratory for Diagnosis and Treatment of Brain Diseases, College of Pharmaceutical Sciences, Soochow University, Suzhou, Jiangsu 215123, China.
8. MOE Key Laboratory of Geriatric Diseases and Immunology, Suzhou Medical College of Soochow University, Suzhou, Jiangsu 215123, China.
9. Suzhou Key Laboratory of Geriatric Neurological Disorders, the First People's Hospital of Taicang, Taicang Affiliated Hospital of Soochow University, Suzhou, Jiangsu, 215400, China.
Received 2025-10-22; Accepted 2026-5-4; Published 2026-5-18
Abstract

Ubiquitin-fold modifier 1 (UFM1) covalently modifies protein substrates (UFMylation) and alters their biological functions. Genetic screening disclosed that enzymes in the UFMylation system play critical roles in regulating autophagy. However, it is still elusive which protein is UFMylated and how this modification modulates autophagy. Here, our quantitative proteomics and biochemical experiments identify SQSTM1/p62 as a UFMylation substrate and discover its two major UFMylation sites, K420 and K435. Mutating them to Arg (p622KR) completely abolishes the effect of p62 on autophagic activity. Fusion of UFM1ΔC4 to p622KR (p622KR-UFM1ΔC4) restores the p62-mediated pathogenic autophagic degradation in primary cortical neurons and Huntington's disease mouse striatum. Mechanistically, p62 UFMylation enhances its interaction with LC3, augments autophagic flux, and eliminates pathogenic mutant huntingtin. Collectively, this work discovers a new post-translational modification, UFMylation, on p62 and establishes this modification as a key regulator of autophagy that promotes the clearance of mutant huntingtin, offering a potential target for therapeutic intervention.
Keywords: SQSTM1/p62, UFMylation, quantitative proteomics, autophagic degradation, Huntington's disease, mutant huntingtin
Introduction
Autophagy-lysosome pathway (ALP) is one of the cellular protein degradation modalities in eukaryotes, which sustains cellular homeostasis by degrading unfolded, misfolded, or unwanted proteins [1]. Typically, the ALP acts primarily to remove protein aggregates, damaged organelles, and intracellular pathogens through the formation of autophagosomes that are subsequently fused with lysosomes for proteolysis. Dysfunction of the ALP is associated with many diseases, including neurodegenerative diseases, cancer, microbial infection, and immune disorders [2-5]. Although it has been discovered that autophagy is modulated both at the transcriptional and post-translational levels, the regulatory mechanism of the ALP is not completely understood.
The pathogenic hallmark of Huntington's disease (HD) is the aberrant expansion of the CAG trinucleotide repeats in the huntingtin (HTT) gene. This neurodegenerative disorder is manifested as progressive degeneration in the striatum and cortex, marked by the gradual loss of medium spiny neurons (MSNs) alongside pyramidal neurons, ultimately impairing cortico-striatal circuitry [6]. Emerging studies on the pathogenesis of HD have demonstrated that autophagosome-tethering compounds effectively facilitate the clearance of mutant HTT (mHTT) by selectively targeting protein aggregates to autophagic machinery [7]. This observation suggests that augmenting selective autophagy flux to enhance the clearance of mHTT constitutes a promising HD intervention paradigm, presenting both theoretical importance and translational potential for developing new therapeutics for neurodegenerative diseases.
SQSTM1/p62 (sequestosome 1, hereafter p62) is a critical autophagy receptor that recruits protein aggregates to autophagosomes through its N-terminal Phox1 and Bem1p domain, LC3-interacting region (LIR), and ubiquitin-associated domain (UBA), thereby promoting the lysosomal degradation of intracellular protein aggregates [8-11]. It is well established that p62 acts as a receptor to colocalize with mHTT and mediates its autophagic degradation [12-15]. The function of p62 is precisely controlled by a diverse array of post-translational modifications (PTMs), such as phosphorylation [16, 17], acetylation [18], ubiquitination [19-21], and FAT10ylation [22]. These modifications modulate the autophagic clearance of cargo proteins by orchestrating their affinity to p62, the capacity of target autophagosomes, and the interaction between p62 and LC3.
UFM1 is a ubiquitin-like modifier that has a strikingly conserved tertiary structure resembling ubiquitin [23]. UFM1 can also covalently modify protein substrates (UFMylation) through an enzymatic cascade comprising the UFM1-activating enzyme UBA5, the UFM1-conjugating enzyme UFC1, and the UFM1-protein ligase UFL1 [24, 25]. Interestingly, the DDRGK domain-containing protein 1 (DDRGK1 or UFBP1) is also an important adaptor that promotes protein UFMylation [26]. In addition, conjugated UFM1 could be removed by UFSP1 and UFSP2, two UFM1-specific proteases [27-29]. Besides the components in the UFMylation system, more than two dozen proteins were discovered to be UFMylated [24]. Recent studies have revealed that UFMylation plays diverse roles in many cellular processes, including endoplasmic reticulum (ER) homeostasis [30], protein biogenesis [31], cell cycle progression [32], DNA damage response [33, 34], brain development [35], and cancer-associated signaling pathways [26, 36-39]. Emerging evidence indicates that UFMylation modulates protein degradation through the ALP, highlighting its potential as a therapeutic target. Specifically, VCP/p97 UFMylation at K107 enhances the stabilization of beclin-1 and facilitates the formation of the phosphatidylinositol 3-kinase (PI3K)/PtdIns3K complex. This finding indicates that VCP/p97 UFMylation plays a vital regulatory function in autophagy initiation [40]. In addition, UFL1 interacts with NLRP3 and mediates its UFMylation during activation, while stabilizing the NLRP3 protein by inhibiting K63-linked polyubiquitination and diminishing its subsequent autophagic degradation [41]. However, all of these studies explored the autophagic degradation of UFMylated substrates without delving into the relationship between UFMylation and autophagy.
Previous work revealed that UFL1-deficiency in bone marrow cells attenuated autophagic degradation [42]. Autophagy-related protein 8-family proteins (including LC3 in humans) interact with the non-canonical LIR motif in UBA5, regulating its subcellular distribution and biological functions [43]. Depletion of UFBP1 hampers the autophagosome-lysosome fusion, which results in the accumulation of autophagosomes and enhanced apoptosis in mouse embryonic fibroblasts [44]. CRISPR/Cas9-based genetic screening disclosed that the p62 function was regulated by UFM1 and the components in the UFMylation system [45]. Nonetheless, it is elusive whether p62 is UFMylated, and the precise biochemical mechanisms by which UFMylation regulates autophagy have yet to be elucidated.
Here, we employ tandem affinity purification coupled with quantitative proteomics to identify the potential UFMylated proteins and confirm that p62 is mainly UFMylated at K420 and K435. We further demonstrate that the wild-type (WT) p62 but not the UFMylation-deficient mutant promotes the LC3II accumulation in autophagic degradation upon p62 knockdown in cell lines and in primary cortical neurons. Fusion of a C-terminal truncated UFM1 to the UFMylation-deficient p62 mutant almost completely restores the function of p62 as an autophagy receptor. Mechanistically, p62 UFMylation enhances the binding of p62 to LC3II and thereby promotes autophagic degradation of pathogenic mHTT. Moreover, p62 UFMylation is attenuated in the cortex of transgenic HD mice, and mimicking p62 UFMylation promotes the clearance of mHTT and reinstates the density of MSNs in the striatum of HD mice. Thus, we discover a new PTM, UFMylation, on p62 and reveal a novel function of p62 UFMylation in elevating autophagic activity, offering new insights into developing potential therapeutic strategies for autophagic degradation of pathogenic mHTT by targeting the UFMylation system.
Results
Depletion of UFL1 or UBA5 impairs the autophagic flux
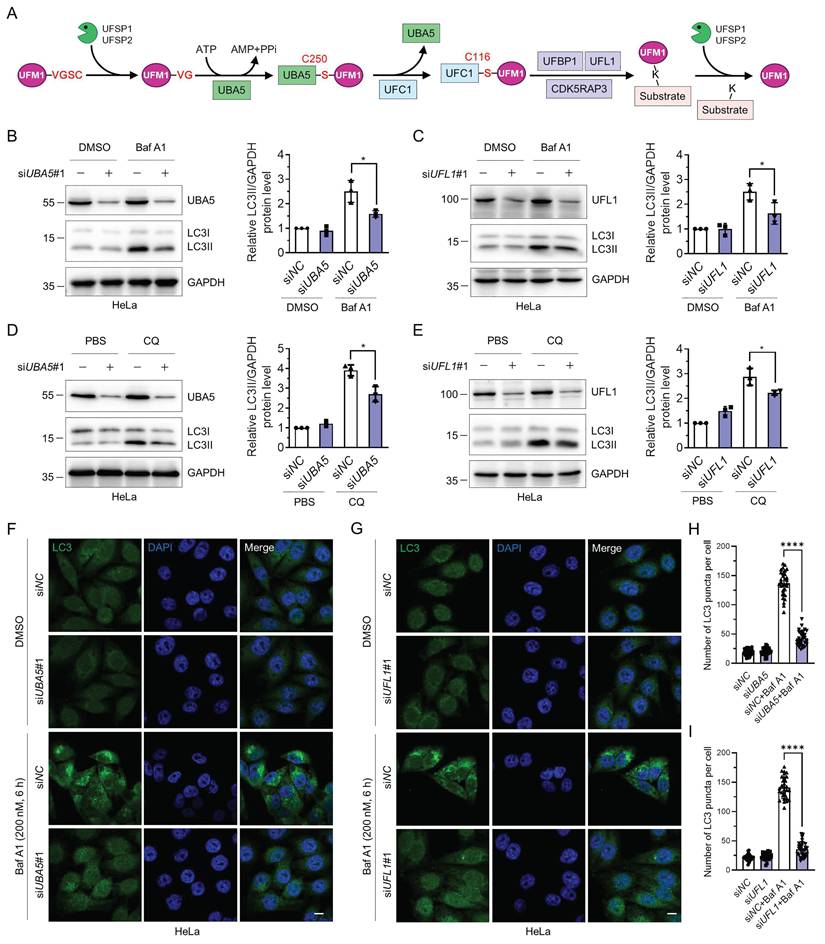
The UFMylation cascade is initiated by cleaving the last two amino acids in UFM1 precursor by UFSP1 and UFSP2. Subsequently, mature UFM1 is covalently linked to target proteins through the UBA5-UFC1-UFL1 tripartite enzymatic reactions. UFBP1 and/or CDK5RAP3 are essential regulators for substrate UFMylation. Furthermore, UFSP1 and UFSP2 regulate the deUFMylation of substrates, rendering UFMylation a reversible modification (Fig. 1A). Previously, it has been reported that UFL1 deficiency impairs autophagic degradation [42]. Atg8-family proteins interact with UBA5 and regulate its localization and biological functions [43]. However, the regulatory roles of UFL1 or UBA5 in autophagy and the underlying molecular mechanisms are still mysterious. Here, we sought to explore the function of UBA5 and UFL1 in autophagic degradation. Knockdown of either UBA5 or UFL1 significantly reduced the LC3II accumulation induced by two late-stage autophagy inhibitors, bafilomycin A1 (Baf A1) and chloroquine (CQ) [46] (Fig. 1B-E), indicating that UBA5 or UFL1 depletion attenuates the autophagic flux. Similar phenomena were observed in MCF-7 cells (Fig. S1A-D). Knockdown of UBA5 or UFL1 with another set of siRNAs (siUBA5#2 and siUFL1#2) also exhibited a similar trend (Fig. S1E-H). Consistently, beclin-1 was also reduced when UBA5 or UFL1 was depleted in HeLa cells (Fig. S1I-J). To further explore the role of these two proteins in the regulation of autophagy, we examined LC3 puncta by immunofluorescence in the control (siNC), UBA5- or UFL1-depleted (siUBA5 or siUFL1) HeLa cells. In the control group, the number of LC3 puncta was increased markedly upon the inhibition of autophagic degradation by Baf A1. However, interestingly, Baf A1-induced increase of LC3 puncta was pronouncedly attenuated in the UBA5- or UFL1-depleted cells, indicating that UBA5 and UFL1 were indeed two general regulators of autophagy (Fig. 1F-I and Fig. S1K-L). Thus, these findings indicate that depletion of UBA5 or UFL1 inhibits autophagic flux.
Knockdown of UBA5 or UFL1 attenuates the autophagic activity. (A) The schematic illustration of the UFMylation pathway. UFM1 is synthesized as a precursor which is hydrolyzed by UFSP1/2 at its C-terminus to expose glycine. The mature UFM1 is then activated and subjected to a nucleophilic attack by the Cys250 of UBA5. Then activated UFM1 is transferred to UFC1 Cys116 through a thioester bond. In this process, UFL1 forms a tripartite complex with UFBP1 or CDK5RAP3 and transfers UFM1 to lysine residues of target proteins. UFSP1/2 mediate the deUFMylation by removing UFM1 from substrates. (B-E) Knockdown of UBA5 or UFL1 reduces autophagic activity. HeLa cells were transfected with siNC, siUBA5#1, or siUFL1#1 for 42 h and treated with bafilomycin A1 (Baf A1, 200 nM for 6 h) (B-C) or chloroquine (CQ, 50 μM for 12 h) (D-E). Cell lysates were immunoblotted for LC3, UBA5, and UFL1. The relative protein levels of LC3II were normalized to GAPDH and quantified. Data were presented as mean ± SD (n = 3 biological replicates), two-way ANOVA with Tukey's multiple comparisons post hoc test, *: P < 0.05. (F-I) LC3 puncta are reduced in UBA5- or UFL1-depleted HeLa cells. Immunofluorescence staining (F-G) and quantification (H-I) of LC3 in HeLa cells treated as in (B-C). Scale bar, 10 μm. Mean ± SD (n = 3 biological replicates with 30 cells, each point represented the number of puncta in one cell), two-way ANOVA with Tukey's multiple comparisons post hoc test, ****: P < 0.0001.
p62 is a bona fide UFMylation substrate and is mainly UFMylated on K420 and K435
To investigate whether UBA5 or UFL1 directly modulates the UFMylation of autophagy-related proteins to regulate autophagic flux, we performed tandem affinity purification and quantitative proteomics analysis of possible UFMylated proteins. We expressed the tagged UFMylation components, including His6-BAP-UFM1, HA-UBA5, Myc-UFC1, Myc-UFL1, and Myc-UFBP1, and BirA* in HEK293T-sgUFSP2 cells, followed by biotin treatment and cell lysis. The UFMylated proteins were first purified with Ni-NTA resin and further enriched with NeutrAvidin agarose resin under denaturing conditions (Fig. 2A). The purification was verified by silver staining and Western blotting (Fig. 2B), followed by in-gel trypsinolysis. The resulting peptides were subjected to LC-MS/MS followed by data analysis. The identified proteins from three biological replicates were quantified to generate the volcano plot. Among the 2473 identified proteins (Table S1), 738 proteins (Table S2) exhibited statistically significant differences and were considered putative UFMylated substrates (Fig. 2C). Many previously discovered UFMylated proteins, including UFBP1, RPL26, Histone H4, p53, P4HB, and PARP1 (Table S2), were also identified, indicating the effectiveness of our high-throughput proteomic approach. Interestingly, among them, p62, a critical autophagy receptor [8], is a novel UFMylation candidate. LC-MS/MS confidently identified 13 unique tryptic peptides derived from p62 with a sequence coverage of 71.14%, one of them bearing the UFMylation site (Fig. 2D). We speculate that UFMylation of p62 may play a critical function in modulating autophagy flux.
Identification and verification of p62 UFMylation by quantitative proteomics and biochemical approaches. (A) Schematic illustration of the experimental procedure for the identification of putative UFMylation substrates. HEK293T-sgUFSP2 cells were transfected with the UFMylation components (including His6-BAP-UFM1, HA-UBA5, Myc-UFC1, Myc-UFL1, and Myc-UFBP1) and BirA* for 24 h, and treated with biotin (20 μM) for 24 h. UFMylated proteins were purified from cell lysates sequentially with Ni-NTA resin and NeutrAvidin agarose under denaturing conditions. The purified UFMylated proteins were separated by SDS-PAGE and digested in gel with trypsin. Peptides were desalted with C18 ZipTip and analyzed by LC-MS/MS followed by database search. (B) Silver staining and immunoblotting of UFM1 for the affinity-purified proteins. (C) A volcano plot for the affinity-purified proteins identified by LC-MS/MS. p62/SQSTM1 was indicated by the open black circle. (D) List of MS-identified tryptic peptides derived from p62. The amino acid sequence, charge state (z), MH+, missed cleavage #, Δmass (ppm), Xcorr, and amino acid position for the peptides were provided. (E) Biochemical validation of p62 as a UFMylation substrate. HEK293T cells were transfected with His6-UFM1, HA-UBA5, Myc-UFC1, Myc-UFL1, Myc-UFBP1, and/or FLAG-p62 for 48 h. p62 was purified with anti-FLAG affinity gel and eluted with FLAG peptide (200 μg/mL). The cell lysates and purified samples were immunoblotted. (F) Tandem affinity purification and immunoblotting of the UFMylated p62. The cell lysates and proteins affinity-purified according to the experimental procedure in Fig. 1A were immunoblotted. (G-H) Endogenous p62 is UFMylated. HCT116 cells (G) and mouse cortex (H) were lysed and endogenous p62 was immunoprecipitated with IgG or anti-p62 antibodies for immunoblotting analysis. (I) UFMylation assay of p62. HEK293T cells were transfected with FLAG-p62, HA-UBA5, Myc-UFC1, Myc-UFL1, Myc-UFBP1, and V5-UFM1/V5-UFM1ΔC2/V5-UFM1ΔC3 for 48 h. The UFMylation assay was performed as described in (E). *: non-specific signal.
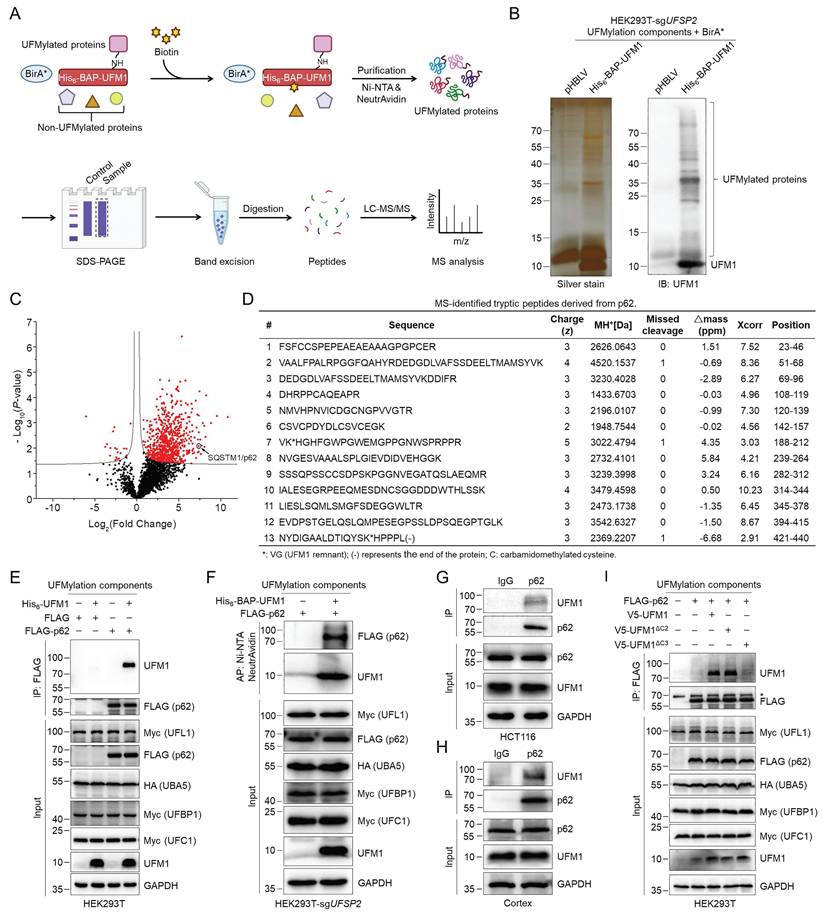
To verify whether p62 is a target for UFMylation biochemically, we co-expressed FLAG-p62 with the UFMylation components in HEK293T cells and isolated p62 using FLAG affinity gel. Immunoblotting of the purified p62 with an anti-UFM1 antibody detected a band at a molecular weight approximately equal to the sum of p62 and UFM1 (Fig. 2E). To rule out the possible noncovalent interaction of UFMylated proteins, we further immunoblotted the tandem affinity-purified samples obtained under denaturing conditions based on the procedure described in Fig. 2A from cells expressing FLAG-p62 and the UFMylation components. The result explicitly demonstrated that p62 migrated to a higher molecular weight region with the addition of a UFM1 molecule, confirming that UFM1 was covalently conjugated to p62 (Fig. 2F). The anti-UFM1 immunoblotting of immunoprecipitated endogenous p62 from HCT116 cells and mouse cortex also detected a clear band at the similar molecular weight, indicating that the endogenous p62 was indeed UFMylated (Fig. 2G-H). Our experiments further revealed that p62 can be mono-UFMylated via expressing either premature (UFM1) or mature UFM1 (UFM1ΔC2), but not the conjugation-defective UFM1 mutant (UFM1ΔC3) (Fig. 2I). Based on the above experiments, we could confidently conclude that p62 is indeed a bona fide UFMylation substrate.
We next sought to identify the UFMylation sites on p62. Since the quantitative proteomics identified one potential UFMylation site (K435) on p62 with high confidence (Fig. S2), and K435 is located near the C-terminal tail of p62, which usually collaborates with K420 in contributing to the function of the p62 UBA domain, we generated several p62 K-to-R mutants and performed the UFMylation assay. The results demonstrated that the K420R and K435R mutations reduced the p62 UFMylation, and the K420/435R p62 double mutant (referred to as p622KR) can hardly be UFMylated (Fig. 3A). The UFMylation of p62 was further confirmed in vitro (Fig. 3B). Purified GST-p62 and GST-p622KR were incubated in the UFMylation buffer containing UBA5, UFC1, UFL1, UFM1ΔC2, ATP, and MgCl2. The reaction was stopped by adding SDS sample loading buffer, and the samples were subjected to immunoblotting with anti-UFM1 antibody. The result demonstrated that p62 was clearly UFMylated (lane 10) while p622KR was barely UFMylated (lane 11), indicating that K420 and K435 are the major UFMylation sites on p62. Additionally, we examined the UFMylation of p62 in cortical tissues from mice at different ages and found that UFMylation of p62 gradually decreased as age increased, suggesting that p62 UFMylation may be age-dependent (Fig. S3A-B).
Biochemical identification of p62 UFMylation sites and knockdown of UBA5 or UFL1 attenuates p62 UFMylation. (A) K420 and K435 are the major UFMylation sites on p62. HEK293T cells were transfected with the wild-type (WT) p62 or its K-to-R mutants, enzymes in the UFMylation system, and His6-UFM1. The UFMylation assay was performed as described in Fig. 2E. The relative intensity for the p62 UFMylation was depicted below the image. (B) In vitro p62 UFMylation. Recombinant UFM1 conjugation components (UFM1ΔC2, UBA5, UFC1, UFL1, UFBP1) and GST-p62 or GST-p622KR were incubated in the reaction buffer at 30 °C for 2 h. The reaction was terminated by adding 5 × SDS sample loading buffer and heating at 98 ºC for 10 min, and the samples were immunoblotted with the indicated antibodies. (C) UFSP2 knockout enhances p62 UFMylation. The UFMylation assay was performed in the UFSP2 knockout HEK293T cells as described in Figure 2E. (D) Effect of UBA5, UFC1, UFL1, and UFBP1 on p62 UFMylation. HEK293T cells were transfected with different combinations of the UFM1 conjugation components for 48 h and the UFMylation assay was performed as described in Fig. 2E. (E-F) p62 UFMylation is decreased after knocking down UBA5 (E) or UFL1 (F). UFSP2 knockout HEK293T cells were transfected with siNC, siUBA5#1, or siUFL1#1 along with FLAG-p62 and His6-UFM1 plasmids for 48 h. p62 was immunoprecipitated with anti-FLAG affinity gel. The cell lysates and immunoprecipitates were immunoblotted.
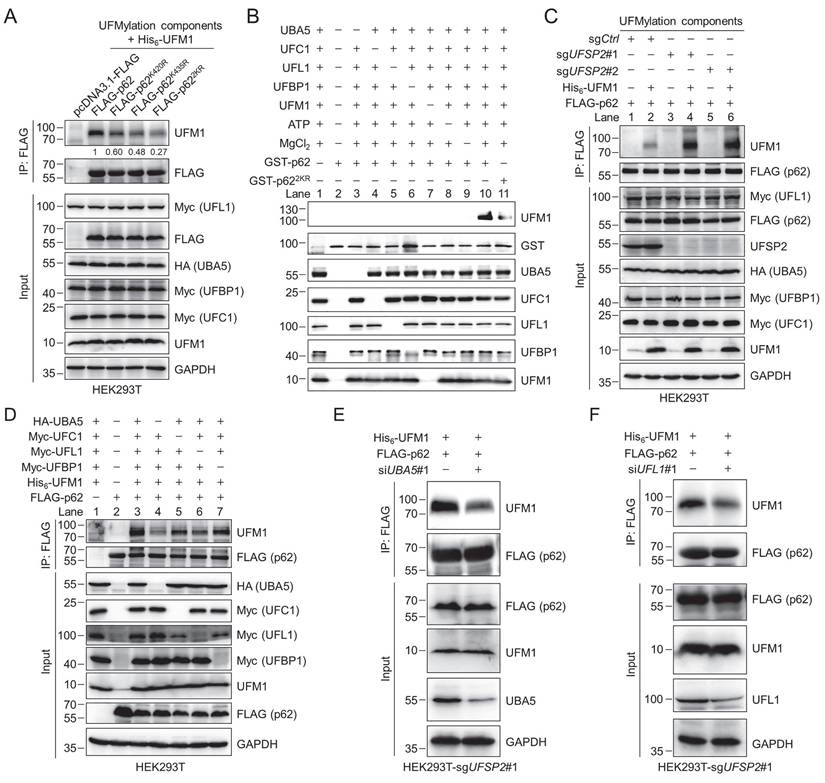
To further exploit the function of each component in the UFMylation system, we conducted the following experiments. When we overexpressed p62 and the UFMylation components in the HEK293T-sgUFSP2 cells, the p62 UFMylation level was strikingly elevated compared with the sgCtrl cells (lanes 4 and 6) (Fig. 3C). Furthermore, p62 UFMylation was markedly reduced when UBA5 (lane 4) or UFL1 (lane 6) was not ectopically expressed. However, UFMylated p62 could be readily detected when cells did not express exogenous UFC1 (lane 5) or UFBP1 (lane 7) (Fig. 3D). Notably, after depletion of either UBA5 or UFL1 by specific siRNAs, the p62 UFMylation level was diminished (Fig. 3E-F). These results indicate that UBA5 and UFL1 are important, at least under our experimental conditions, for p62 UFMylation, presumably due to their insufficient endogenous expression in this cell line, and UFSP2 can remove the conjugated UFM1 from p62.
p62 UFMylation modulates autophagosome clearance by elevating autophagic flux
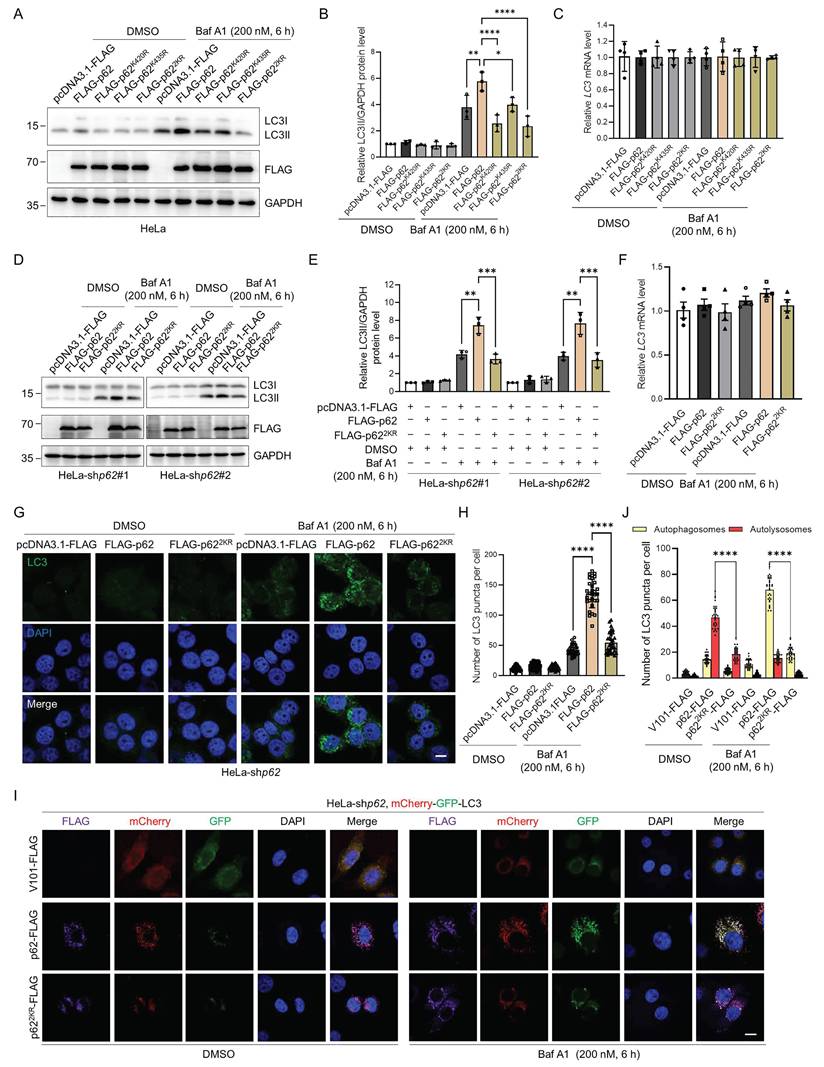
p62 is a critical autophagy receptor and could be modified by an array of PTMs that modulate its autophagic activity [16-22, 47-49]. Therefore, we speculated that p62 UFMylation might also affect its autophagic activity. Consistent with this hypothesis, when p62 was overexpressed, LC3II accumulated upon Baf A1 treatment due to the blockage of lysosomal degradation. Strikingly, expression of the UFMylation-deficient p62 mutant significantly attenuated this Baf A1-induced LC3II accumulation without affecting its mRNA level. This indicates that impairment of p62 UFMylation diminishes autophagic flux (Fig. 4A-C and Fig. S4A-C). To exclude the impact of endogenous p62, we examined the function of p62 UFMylation in p62 knockdown HeLa cells (Fig. S4D). Consistent with the above results, WT p62 but not UFMylation-deficient p62 promoted LC3 accumulation, whereas the LC3 mRNA level was not altered by p62 mutation (Fig. 4D-F). Additionally, immunostaining of LC3 puncta further confirmed this finding. As expected, WT p62 elevated while p622KR failed to increase the number of LC3II puncta in p62 knockdown HeLa cells treated with Baf A1 (Fig. 4G-H). To further evaluate the autophagic flux level, we used tandem-tagged mCherry-GFP-LC3 [50] to distinguish autophagosomes and autolysosomes. After autophagosomes fuse with lysosomes, autolysosomes become acidic, leading to the reduction of the GFP fluorescence signal due to its reduced sensitivity to low pH, and thus, only the mCherry (red) fluorescence signal could be detected. Furthermore, Baf A1 treatment resulted in the detection of mCherry-GFP (yellow), which was due to the blockage of the autophagosome-lysosome fusion. Although Baf A1 treatment may increase lysosomal pH and partially restore GFP signal, the results were obtained based on the relative changes under identical experimental conditions. To further validate the results, we overexpressed the WT or UFMylation-deficient p62 mutant p622KR in p62 knockdown HeLa cells and examined the intracellular autophagic flux upon DMSO or Baf A1 treatment. Detailed analysis revealed that the number of autolysosomes (red) was markedly increased in cells expressing the WT p62, but it was only marginally increased in the p622KR expressing cells upon DMSO treatment. Moreover, autophagosomes (yellow) were significantly elevated in cells expressing the WT p62 but not in cells expressing p622KR in response to Baf A1 (Fig. 4I-J, and Fig. S4E). Taken together, these data suggest that p62 UFMylation modulates autophagosome formation and thus elevates autophagic flux.
p62 UFMylation is required for LC3II accumulation in autophagic degradation. (A-B) UFMylation-deficient p62 mutation abolishes the effect of p62 on autophagy. HeLa cells were transfected with pcDNA3.1-FLAG, FLAG-p62, FLAG-p62K420R, FLAG-p62K435R, or FLAG-p622KR plasmid for 42 h, and then treated with Baf A1 (200 nM) for 6 h. Cell lysates were subjected to immunoblotting analysis. The relative protein levels of LC3II were normalized to GAPDH and quantified. Mean ± SD (n = 3 biological replicates), two-way ANOVA with Tukey's multiple comparisons post hoc test, *: P < 0.05, **: P < 0.01, ****: P < 0.0001. (C) Mutation of the p62 UFMylation sites does not affect the LC3 mRNA level. Total RNA was isolated from HeLa cells treated as described in (A) and subjected to RT-qPCR analysis. (D-E) The absence of UFMylation in p62 eliminates the regulatory role of p62 on autophagy. p62 knockdown HeLa cells were transfected with pcDNA3.1-FLAG, FLAG-p62, or FLAG-p622KR plasmid for 42 h, and then treated with Baf A1 (200 nM) for 6 h. Cell lysates were subjected to immunoblotting analysis (D) and quantification (E). The relative protein levels of LC3II were normalized to GAPDH and quantified. Mean ± SD (n = 3 biological replicates), two-way ANOVA with Tukey's multiple comparisons post hoc test, **: P < 0.01, ***: P < 0.001. (F) UFMylation-deficient p62 mutation does not affect the LC3 mRNA level in p62 knockdown HeLa cells. Total RNA was isolated from p62 knockdown HeLa cells treated as described in (D) and subjected to RT-qPCR analysis. Mean ± SD (n = 3 biological replicates), two-way ANOVA with Tukey's multiple comparisons post hoc test. (G-H) UFMylation-deficient mutation eliminates the influence of p62 on the formation of LC3 puncta in p62 knockdown HeLa cells. p62 knockdown HeLa cells were transfected with pcDNA3.1-FLAG, FLAG-p62, or FLAG-p622KR plasmid for 42 h, and then treated with Baf A1 (200 nM) for 6 h for immunofluorescence imaging (G) and quantification (H). Scale bar, 10 μm. Mean ± SD (n = 3 biological replicates with 30 cells, each point represented the number of puncta in one cell), two-way ANOVA with Tukey's multiple comparisons post hoc test, ****: P < 0.0001. (I-J) p62-enhanced autophagic flux is blocked by UFMylation-deficient p62 mutation. p62 knockdown HeLa cells were transfected with the mCherry-GFP-LC3 plasmid and V101-FLAG, p62-FLAG, or p622KR-FLAG plasmid, and then treated with DMSO or Baf A1 (200 nM) for 6 h for confocal imaging (I) and quantitative analysis (J) of autophagosomes (yellow puncta) and autolysosomes (red puncta). Scale bar, 10 μm. Mean ± SD (n = 3 biological replicates with 30 cells, each point represented the number of puncta in one cell), two-way ANOVA with Tukey's multiple comparisons post hoc test, ****: P < 0.0001.
Since p622KR exhibited different autophagic activity compared with WT p62, we hypothesized that the regulatory function of p62 might be highly influenced by UFMylation. To further investigate the function of p62 UFMylation, we constructed an artificial p622KR-UFM1ΔC4 mutant to mimic p62 UFMylation by fusing UFM1ΔC4 (UFM1 with deleted C-terminal 4 amino acids VGSC) to the C-terminus of p622KR, because the major UFMylation sites on p62 are close to its C-terminus (Fig. 5A). This strategy was previously used to study the function of ISGylation and SUMOylation [51, 52]. Immunoblotting experiments revealed that LC3II accumulation was increased when WT p62 or p622KR-UFM1ΔC4, but not p622KR, was expressed in HeLa cells, followed by Baf A1 treatment (Fig. 5B-C). As expected, immunostaining unveiled that LC3 puncta exhibited similar phenomena as the immunoblotting result, i.e., expression of p622KR-UFM1ΔC4 significantly restored the number of LC3 puncta in p62 knockdown HeLa cells in the presence of Baf A1 (Fig. 5D-E). We then exploited the effect of p62 UFMylation on LC3 puncta in primary cortical neurons. Unsurprisingly, immunoblotting and immunofluorescence experiments uncovered that expression of p62 or p622KR-UFM1ΔC4 but not p622KR recovered LC3II accumulation (Fig. 5F-I) in primary cortical neurons.
p62 UFMylation modulates autophagosome clearance by elevating autophagic flux. (A) Schematic representation of UFM1, p62, p622KR, and p622KR-UFM1ΔC4. UFM1ΔC4 was fused to the C-terminus of p622KR to mimic the UFMylated p62 because the major UFMylation sites were at the C-terminus of p62. (B-C) p62 UFMylation elevates LC3II protein level. HeLa cells were transfected with pcDNA3.1-FLAG, FLAG-p62, FLAG-p62K420R, FLAG-p62K435R, FLAG-p622KR, FLAG-p62K420R-UFM1ΔC4, FLAG-p62K435R-UFM1ΔC4, or FLAG-p622KR-UFM1ΔC4 plasmid, and then treated with Baf A1 (200 nM) for 6 h. Cell lysates were subjected to immunoblotting analysis. The relative protein levels of LC3II were normalized to GAPDH and quantified. Mean ± SD (n = 3 biological replicates), one-way ANOVA with Tukey's multiple comparisons post hoc test, *: P < 0.05, **: P < 0.01, ***: P < 0.001. (D-E) p62 UFMylation increases LC3 puncta. p62 knockdown HeLa cells were transfected with pcDNA3.1-FLAG, FLAG-p62, FLAG-p622KR, or FLAG-p622KR-UFM1ΔC4 plasmid, and then treated with Baf A1 (200 nM) for 6 h. Cells were immunostained with LC3 and DAPI (D) for quantification (E). Scale bar, 10 μm. Mean ± SD (n = 3 biological replicates with 30 cells, each point represented the number of puncta in one cell), two-way ANOVA with Tukey's multiple comparisons post hoc test, ****: P < 0.0001. (F-G) p62 UFMylation elevates LC3II protein level in primary cortical neurons. Primary cortical neurons were cultured with conditioned medium for 5 days and infected with pHBLV-FLAG, p62-FLAG, p622KR-FLAG, or p622KR-UFM1ΔC4-FLAG lentiviral particles for 42 h, and then treated with DMSO or Baf A1 (200 nM) for 6 h. Cell lysates were subjected to immunoblotting analysis (F) and quantification (G). The relative protein levels of LC3II were normalized to β-actin and quantified. Mean ± SD (n = 3 biological replicates), two-way ANOVA with Tukey's multiple comparisons post hoc test, *: P < 0.05, **: P < 0.01, ns: not significant. (H) UFMylation-deficient p62 mutation does not affect the LC3 mRNA level in primary cortical neurons. Total RNA was isolated from primary cortical neurons treated as described in (F) and subjected to RT-qPCR analysis. Mean ± SD (n = 4 biological replicates), two-way ANOVA with Tukey's multiple comparisons post hoc test. (I) UFMylation-deficient mutation abolishes the effect of p62 on autophagy in primary cortical neurons. Primary cortical neurons were infected with V101-FLAG, p62-FLAG, p622KR-FLAG, or p622KR-UFM1ΔC4-FLAG lentiviral particles for 42 h, treated with DMSO or Baf A1 (200 nM) for 6 h, and then stained with FLAG, LC3, MAP2, and DAPI. Scale bar, 50 μm. (J-K) p62 UFMylation enhances autophagic flux. p62 knockdown HeLa cells were transfected with the mCherry-GFP-LC3 plasmid and V101-FLAG, p62-FLAG, p622KR-FLAG, or p622KR-UFM1ΔC4-FLAG plasmid for 42 h and then treated with Baf A1 (200 nM) for 6 h for confocal imaging (J) and quantitative analysis (K) of autophagosomes (yellow puncta) and autolysosomes (red puncta). Scale bar, 10 μm. Mean ± SD (n = 3 biological replicates with 30 cells, each point represented the number of puncta in one cell), two-way ANOVA with Tukey's multiple comparisons post hoc test, ****: P < 0.0001. (L-M) p62 UFMylation partially restores the number of autophagic vacuoles. Transmission electron microscopy was carried out for cells transfected with pcDNA3.1-FLAG, FLAG-p62, FLAG-p622KR, or FLAG-p622KR-UFM1ΔC4 plasmid and treated with Baf A1 (200 nM) for 6 h. Representative autophagic vacuoles were indicated by arrows (L) and quantified (M). Scale bar, 500 nm. Mean ± SD (n = 5 biological replicates), one-way ANOVA with Tukey's multiple comparisons post hoc test, *: P < 0.05, **: P < 0.01.
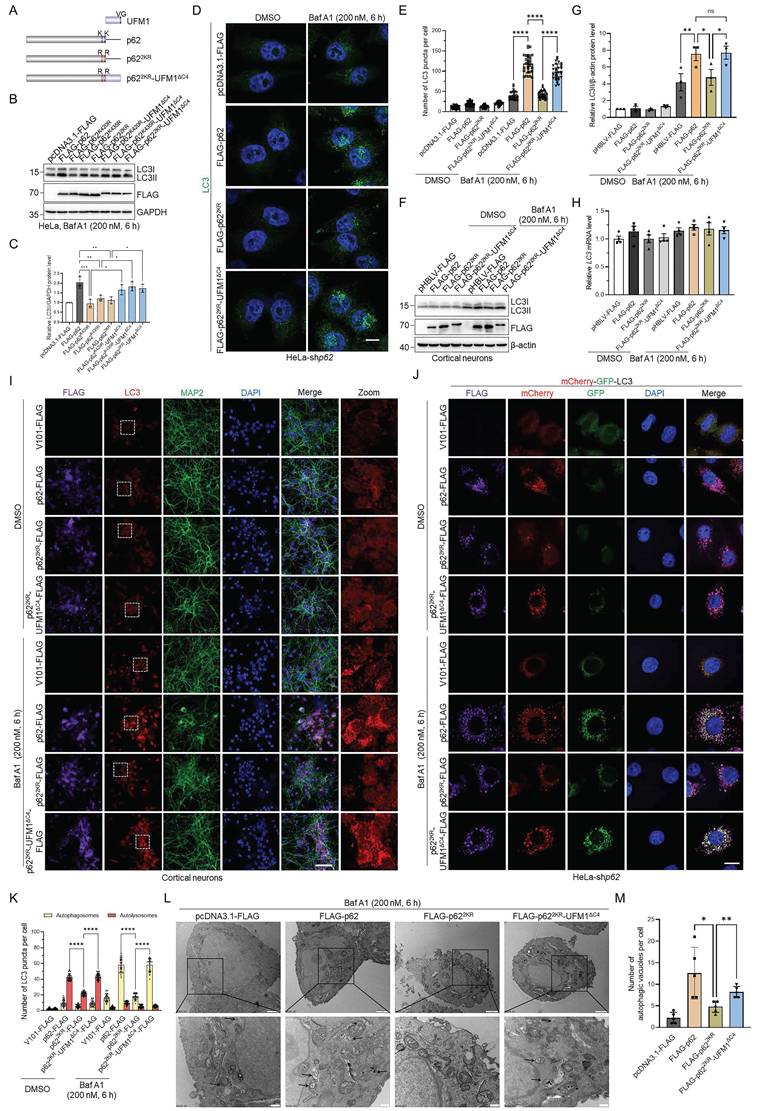
In addition, autophagic flux was blocked upon expressing p622KR, whereas p622KR-UFM1ΔC4 notably restored the autophagosome formation, indicating that p62 UFMylation enhanced autophagic flux in p62 knockdown HeLa cells (Fig. 5J-K and Fig. S4F). In accordance with this, transmission electron microscopy disclosed that p62 UFMylation resulted in a partial restoration of autophagosomes (Fig. 5L-M). Cumulatively, these data underscore that p62 UFMylation plays a crucial role in regulating p62-mediated autophagy.
p62 UFMylation promotes the p62-LC3II interaction and the degradation of pathogenic mHTT
In above experiments, we observed an intriguing phenomenon that p62 UFMylation modulates autophagosome formation and thus elevates autophagic flux. To investigate the molecular mechanisms by which p62 UFMylation modulates its biological functions, we performed co-immunoprecipitation experiments. We expressed His6-Ub and/or UFM1 with FLAG-p62 or GFP-p62 in the UFSP2 knockout HEK293T cells and then examined the self-oligomerization of p62 and its association with ubiquitinated proteins, given that both p62 dimerization and its binding to ubiquitinated cargos are critical for autophagic flux. Surprisingly, the results revealed that UFM1 affected neither the dimerization of p62 nor its interaction with ubiquitinated proteins (Fig. S5A-B). However, unexpectedly, expression of UFM1 increased the p62 UFMylation and promoted the binding between p62 and LC3 (Fig. 6A). In contrast, p622KR weakened the interaction between p62 and LC3 while p622KR-UFM1ΔC4 restored this interaction (Fig. 6B). Along with the above data, these results clearly suggest that UFMylation enhances p62 autophagic activity by increasing the binding of p62 to LC3.
p62 UFMylation promotes the interaction between p62 and LC3 and enhances the degradation of pathogenic mutant huntingtin (mHTT). (A-B) p62 UFMylation enhances the interaction between p62 and LC3. HEK293T-sgUFSP2 cells were transfected with FLAG-p62 and/or His6-UFM1 plasmids (A) or with pcDNA3.1-FLAG, FLAG-p62, FLAG-p622KR, or FLAG-p622KR-UFM1ΔC4 and His6-UFM1 plasmids (B) for 48 h. Cells were lysed and p62 was purified with the anti-FLAG antibody. Cell lysates and affinity-purified samples were immunoblotted. (C-F) p62 UFMylation promotes the degradation of pathogenic mHTT. p62 knockdown HeLa cells were transfected with pcDNA3.1-FLAG, FLAG-p62, FLAG-p622KR or FLAG-p622KR-UFM1ΔC4, and GFP-Htt60Q plasmids for 48 h. Representative confocal images (C) were captured for quantification of GFP-Htt60Q (D). The cell lysates were immunoblotted with the indicated antibodies (E) for quantitative analysis of GFP-Htt60Q protein (F). Scale bar, 10 μm. Mean ± SD (n = 3 biological replicates with 20 cells, each point represented the number of puncta in one cell), one-way ANOVA with Tukey's multiple comparisons post hoc test, *: P < 0.05, ****: P < 0.0001, ns: not significant. (G-J) p62 UFMylation promotes the degradation of pathogenic mHTT in primary cortical neurons. Primary cortical neurons were infected with mCherry-Htt60Q and V101-FLAG, p62-FLAG, p622KR-FLAG, or p622KR-UFM1ΔC4-FLAG lentiviral particles for 48 h for staining of FLAG, MAP2, and DAPI in primary cortical neurons (G) and quantification of mCherry-Htt60Q (H). Lysates from primary cortical neurons were immunoblotted with the indicated antibodies (I) for quantitative analysis of mCherry-Htt60Q protein (J). Scale bar, 20 μm. Mean ± SD (n = 3), one-way ANOVA with Tukey's multiple comparisons post hoc test, **: P < 0.01, ***: P < 0.001, ns: not significant.
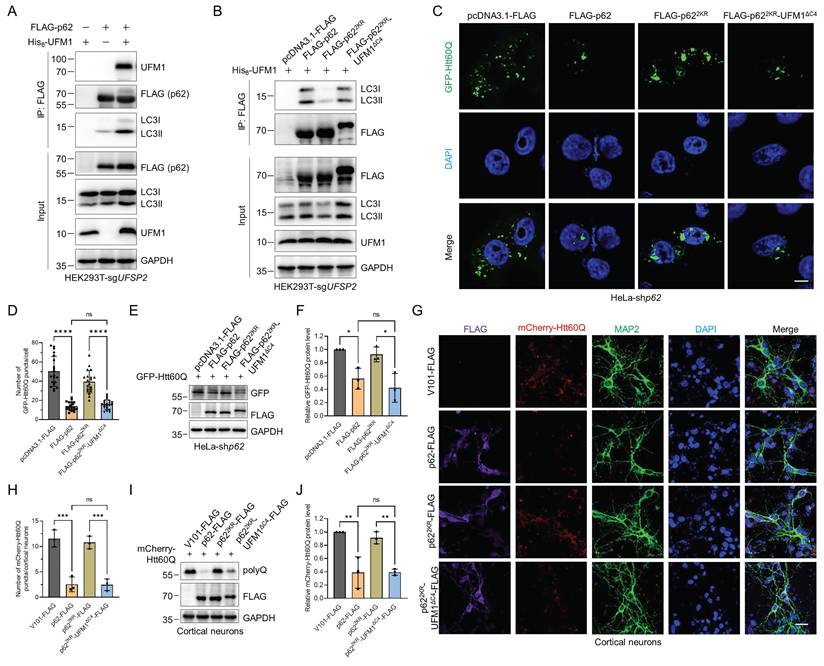
The finding that p62 UFMylation increased the p62-LC3 interaction suggests a regulatory role of UFMylation in autophagic degradation. Therefore, we tested whether p62 UFMylation affects the degradation of neurodegeneration-associated proteins such as pathogenic HTT mutant GFP-Htt60Q, which fuses a GFP to the N-terminal 1-208 amino acids of HTT with a 60-glutamine tail [53]. The results revealed that WT p62, but not p622KR, induced autophagic degradation of Htt60Q. Consistently, the p62 UFMylation mimetic variant p622KR-UFM1ΔC4 also enhanced the degradation of Htt60Q in p62 knockdown HeLa cells (Fig. 6C-F) and primary cortical neurons (Fig. 6G-J). Additionally, overexpression of p62 or its UFMylation mimetic variant p622KR-UFM1ΔC4 promoted the colocalization of LC3 with Htt60Q. Conversely, expression of p622KR reduced their colocalization (Fig. S6A-C). To determine whether the UFMylation of p62 specifically regulates substrate degradation, we examined the interaction between GFP-Htt60Q and WT p62, p622KR, or p622KR-UFM1ΔC4. Immunoprecipitation and immunoblotting experiments revealed that the UFMylation of p62 did not alter its binding affinity for GFP-Htt60Q (Fig. S7A). Consistently, both WT p62 and the p622KR-UFM1ΔC4 promoted the degradation of other neurodegeneration-associated proteins, such as GFP-TauP301L, RFP-SOD1G85R, RFP-SOD1G93A, whereas the p622KR did not affect their degradation (Fig. S7B-D). These data demonstrate that UFMylation increases the interaction between p62 and LC3 and promotes the autophagic degradation of neurodegeneration-associated proteins.
p62 UFMylation promotes the degradation of pathogenic mHTT in HD mice
To comprehensively evaluate the regulatory function of p62 UFMylation in the pathogenesis of HD, we assessed the UFMylation levels of endogenous p62 in the YAC128 and B6-hHTT130-N transgenic mice [54-57]. Affinity purification and immunoblotting results disclosed a significant decline in p62 UFMylation in the cortex of HD mice compared to the WT mice (Fig. 7A and Fig. S8). We subsequently sought to determine whether p62 UFMylation modulates the clearance of mHTT protein in the B6-hHTT130-N mouse striatum. The B6-hHTT130-N mouse carries an N-terminal fragment of the human HTT gene with a 130 CAG repeat expansion, and has been extensively used to study mHTT turnover and degradation mechanisms.
p62 UFMylation enhances the degradation of pathogenic mHTT in the cortex of HD mice. (A) p62 UFMylation is decreased in the cortex of HD mice. The cortex of WT or YAC128 mice was lysed and endogenous p62 was immunoprecipitated with IgG or anti-p62 antibodies for immunoblotting analysis. (B) Schematic illustration of stereotaxic viral injection. B6HTT-CAG130-N mice (8 weeks) were stereotaxically injected with adeno-associated virus in the striatum (coordinates anteroposterior = -1.0 mm, mediolateral = ±2.0 mm, dorsoventral = -3.3 mm). At 12 weeks after injection, the mice were anesthetized and perfused with ice-cold PBS and 4% paraformaldehyde for brain isolation. (C) p62 UFMylation promotes the degradation of mHTT protein in HD mice. At 12 weeks after injection of the indicated adeno-associated virus, the striata of mice were collected and lysed for immunoblotting. (D-E) p62 UFMylation increases the density of medium spiny neurons (MSNs). The mouse brains from (B) were cut into 20 μm sections with a freezing microtome, and the slices were immunostained with DARPP32 and DAPI. Representative confocal images were captured using a confocal microscope (D) for quantification (E) of DARPP32-positive neurons. Scale bar, 100 μm. Mean ± SD (n = 6), one-way ANOVA with Tukey's multiple comparisons post hoc test, ****: P < 0.0001, ns: not significant. (F-G) p62 UFMylation promotes the degradation of mHTT protein in the HD mouse striatum. The mouse brain slices from (D) were immunostained with anti-polyQ antibody and DAPI. Images were scanned using a confocal microscope (F) for quantification (G) of positive polyQ staining. Scale bar, 30 μm. Mean ± SD (n = 6), one-way ANOVA with Tukey's multiple comparisons post hoc test, ****: P < 0.0001, ns: not significant.
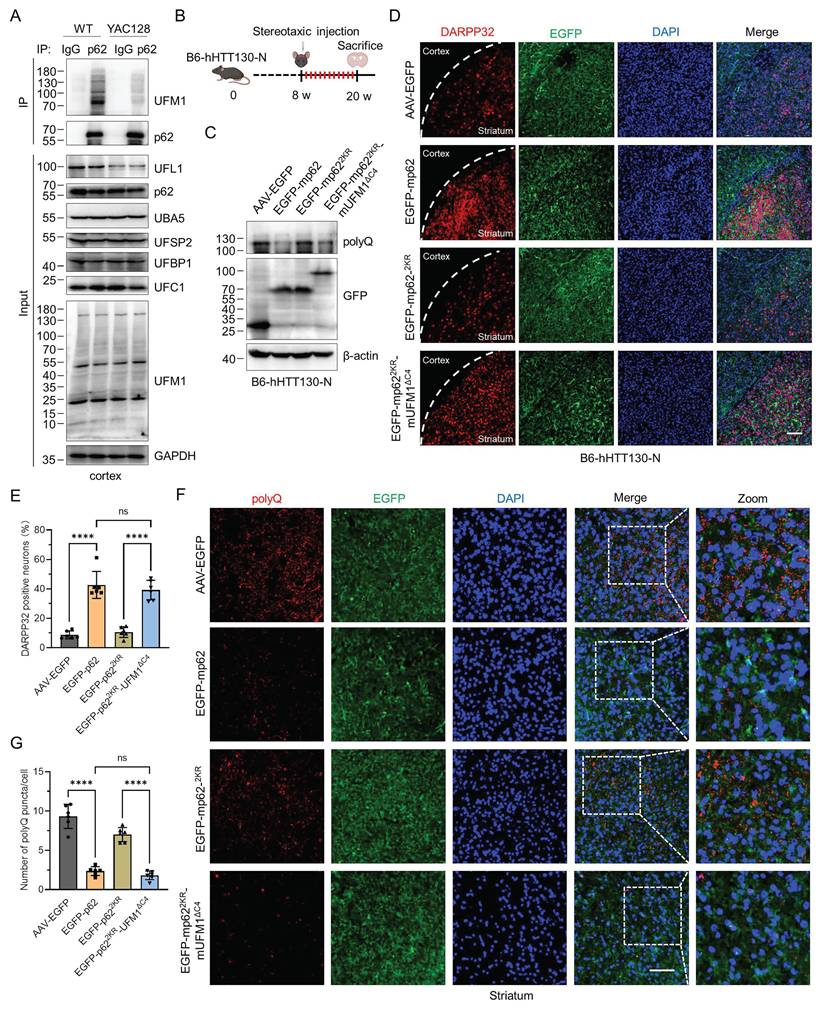
This is because the fragment represents key toxic species implicated in HD pathogenesis and is particularly amenable to assessing alterations in mHTT clearance over relatively short experimental time frames [56]. We generated three distinct murine p62 constructs: FLAG-mp62, UFMylation-deficient mutant FLAG-mp622KR (K422R and K437R), and UFMylation mimetic variant FLAG-mp622KR-mUFM1ΔC4. Immunoblotting coupled with immunoprecipitation confirmed the protein expression and UFMylation of WT and mutant murine p62 (Fig. S9A-B). Then we employed a viral stereotactic injection of recombinant adenoviruses encoding these constructs into the striatum of B6-hHTT130-N mice (Fig. 7B). Analysis of striatal lysate demonstrated that UFMylation deficiency did not promote the clearance of mHTT, whereas mp62 UFMylation mimetic variant decreased the mHTT levels (Fig. 7C). Immunofluorescence results further established that p62 UFMylation preserved the density of MSNs (Fig. 7D-E). In addition, the positive polyQ staining was significantly decreased in the striatum of both AAV9-mp62 and AAV9-mp622KR-mUFM1ΔC4, indicating that p62 UFMylation is capable of augmenting the degradation of mHTT in the striatum of HD mice (Fig. 7F-G). Collectively, these findings demonstrate that UFMylation serves as a critical PTM for p62, which facilitates the clearance of pathogenic mHTT in HD mice.
Discussion
The autophagy-lysosome pathway is the major pathway to remove long-lived proteins, insoluble aggregates, stress granules, and dysfunctional organelles within eukaryotic cells [58]. In this pathway, p62 is a classical receptor that recruits cargos to form autophagosomes and subsequently fuses with lysosomes for degradation [8]. The function of p62 could be precisely controlled by phosphorylation, acetylation, ubiquitination, and FAT10ylation in response to a number of intracellular stimuli [16-22, 47-49]. Here, our affinity purification and quantitative proteomics identifies p62 as a bona fide substrate of UFMylation, which substantially expands the pleiotropic functions of p62 on autophagy. Biochemical experiments discovered that two UFMylation components, UBA5 and UFL1, were important for p62 UFMylation and autophagic degradation. Mapping of the modification sites revealed that K420 and K435 were two major UFMylation sites on human p62. Deficiency of UFMylation on p62 (p622KR) abolished the enhanced effect of p62 on autophagic degradation, while fusion of UFM1ΔC4 to p622KR (p622KR-UFM1ΔC4) significantly recovered the function of p62 upon autophagic inhibition. Mechanistic studies demonstrated that p62 UFMylation affected neither its dimerization nor its recruitment of ubiquitinated proteins. Instead, p62 UFMylation enhanced the binding of p62 to LC3, thereby promoting the autophagic degradation of pathogenic mHTT. Meanwhile, p62 UFMylation was shown to promote autophagic flux and autophagic degradation of mHTT in mouse primary cortical neurons and the striatum of HD mice. Taken together, our work demonstrated that UFMylation of p62 at specific sites promoted its interaction with LC3 and thus recruited mHTT to form mature autophagolysosomes for their degradation. However, disruption of p62 UFMylation impairs the maturation of autolysosomes, resulting in the accumulation of empty autophagolysosomes. Consequently, pathogenic proteins are accumulated, leading to dysfunction in the degradation of cargo proteins or damaged organelles through the ALP (Fig. 8). Coincidently, our proposed mechanism aligns with the observation of empty autophagolysosomes in the brains of HD patients and YAC128 mice, providing a mechanistic explanation for previously reported p62-associated autophagic defects in HD [12, 14].
A proposed model for the regulation of autophagic degradation by p62 UFMylation. UFMylation of p62 enhances autophagy by promoting its interaction with LC3. In a Huntington's disease mouse model, p62 facilitates the degradation of mutant huntingtin (mHTT) protein via UFMylation and increases the number of medium spiny neurons (MSNs) in the striatum.
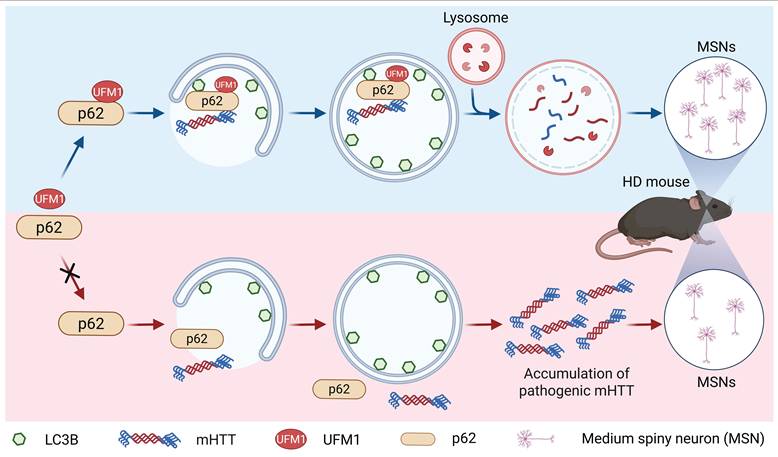
Accumulating evidence indicates that impairment of selective autophagy is highly correlated with the development of neurodegenerative diseases and that enhancing this pathway can confer neuroprotective effects [5, 59]. Recent studies have demonstrated that pharmacological strategies designed to selectively degrade mHTT by autophagy alleviate disease phenotypes in HD mouse models [7]. These findings underscore the importance of efficient cargo recognition and delivery in selective autophagy during neurodegeneration. Consistent with this concept, our study identifies UFMylation of p62 as a regulatory modification that facilitates the clearance of mHTT. Notably, we observed a reduction in p62 UFMylation levels in the cortex of HD model mice, suggesting that dysregulation of UFMylation may be linked to disease-associated defects in selective autophagy. From a mechanistic perspective, we further show that expression of a p62 UFMylation-mimetic variant significantly decreases the accumulation of polyQ in the mouse striatum, supporting the function of p62 UFMylation in limiting the formation of toxic protein aggregation in vivo. UFMylated p62 may reduce mHTT aggregates by degrading non-aggregated mHTT. However, how UFMylation of p62 enhances aggregate clearance and functional improvement in animals remains to be further elucidated. In addition, our results indicate that p62 UFMylation broadly enhances the clearance of diverse aggregation-prone proteins, suggesting that it upregulates autophagic flux rather than acting in a cargo-specific manner.
Although previous genetic screens revealed that components in the UFMylation system regulate autophagy [42-45], it is unknown whether any proteins in the ALP are modified by UFM1 and, if so, what the functions of the modified proteins are. Our quantitative proteomics identified many putatively modified proteins, including previously identified UFMylated proteins such as Histone H4 [33], P4HB [60], p53 [36], and RPL26 [31]. Although tandem MS did not detect modification sites for most proteins, UFMylation sites were identified for RPL26 and all the components in the UFMylation system, such as UBA5, UFC1, UFL1, and UFBP1 (Table S3). Detection of multiple UFMylation sites on UFM1, UFC1, and UFBP1 or two modification sites on one peptide indicates the presence of possible polyUFMylation or multi-mono-UFMylation. These results also indicate that the MS-identified proteins are bona fide or putatively UFMylated proteins. The following reasons might prevent us from identifying the modification sites for the majority of modified proteins. First, some peptides containing UFM1 remnant (Val-Gly) may be too long or too short to be confidently identified by tandem MS. Second, the modified peptides account for only a small fraction of the total peptides analyzed by tandem MS because only one or a few sites in one protein are UFMylated. In this case, the efficiency for the identification of UFMylation sites might be improved if peptides containing the UFM1 remnant could be selectively enriched.
Our work identified the putative UFMylated proteins in cells overexpressing enzymes in the UFM1-conjugation system and biochemically validated the p62 UFMylation. We also discovered the UFMylation of endogenous p62 in cell lines and the mouse cortex. However, the modification patterns were slightly different in these two systems. In human cell lines, we only detected mono-UFMylation for WT p62. It is also possible that the level of polyUFMylation or multi-mono-UFMylation of p62 in these cell lines is much lower than that of the mono-UFMylation, preventing their clear detection on the Western blotting image. However, we detected multiple UFMylated bands after immunoprecipitation of p62 from the mouse cortical tissue although the top bands were much weaker, indicating the presence of multi-mono-UFMylation or polyUFMylation on p62. While we could not tell the exact type of UFMylation for p62 in the mouse cortical tissue, we observed decreased signal for the modified bands in the cortex of aged mice, implying that the UFM1-conjugation system may be dysfunctional in the tissue of aged animals, which is consistent with our discovery that impairment of p62 UFMylation accumulates pathogenic proteins. These data also suggest that the UFM1-conjugation system might vary in cell types, tissues, and organisms.
Although our tandem MS identified p62 K420 bearing the UFM1 remnant (Val-Gly), our biochemical experiments demonstrated that K420 and K435 were the major UFMylation sites. It is likely that a mutation on one site may affect the modification of other sites. This is consistent with the fact that the position for the remaining weak UFMylation of p62 on Western blot after mutating K420 and K435 to Arg is the same as the UFMylated WT p62. These data also indicate that p62 is most likely mono-UFMylated in cells. Although we have identified the major UFMylation sites on p62, it is possible that other modification sites were not detected or detectable by the tandem MS procedure used in this study due to the weak signal or extreme length of the resulting tryptic peptides.
In our experiments, we mutated the major UFMylation sites on p62 to obtain the UFMylation-deficient p62 mutant (p622KR) and then fused the UFM1 with the deleted C-terminal 4 amino acids to this mutant (p622KR-UFM1ΔC4) to mimic the UFMylated p62. Our experiments demonstrated that p622KR lost the function of WT p62 whereas p622KR-UFM1ΔC4 partially or nearly completely recovered the function of WT p62, indicating that p62 UFMylation significantly contributes to the autophagic activity. It should be mentioned that p622KR-UFM1ΔC4 may not completely restore the function of p62 UFMylation for the following possible reasons. First, the p622KR-UFM1ΔC4 fusion protein is not exactly the same as the UFMylated p62 because the chain topology is different from the UFMylated p62 although the major UFMylation sites are located at the p62 C-terminus. Second, UFMylation is a reversible modification that could be eliminated by UFM1-specific proteases. However, p622KR-UFM1ΔC4 cannot be deUFMylated. Third, the identified major UFMylation sites on p62 could also be acetylated or ubiquitinated [18, 21]. Therefore, we could not completely rule out their contribution and the crosstalk between these PTMs and UFMylation on the regulation of autophagic activity. However, it has been demonstrated that the p622KR mutation did not affect the ubiquitination of p62 [18]. Nevertheless, this is one of the best strategies to explore the function of p62 UFMylation.
The activity of p62 is tightly regulated by diverse PTMs. Phosphorylation of p62 at Ser403 by casein kinase 2 and TANK-binding kinase 1 strengthens the interaction between p62 and K63-linked polyubiquitin chains. This modification promotes the formation of sequestosome, thereby enhancing the autophagic clearance of pathogenic protein aggregates [16, 17]. It has been demonstrated that ubiquitination of the UBA domain represents a key regulatory mechanism that determines the binding affinity of p62 for ubiquitinated proteins. However, the p62 UBA domain has a low affinity for ubiquitin and this interaction is further attenuated by its homodimerization [9]. The Keap1/Cullin-3/RBX1 E3 ubiquitin ligase complex mediates the ubiquitination of p62 at K420 in the UBA domain, thereby increasing the interaction between p62 and LC3, enhancing its sequestering activity, and promoting the p62-mediated autophagic degradation [19]. In addition, the ubiquitin-specific protease USP8 selectively removes ubiquitin from K420 on p62 and consequently attenuates the autophagic flux [49]. Moreover, acetylation modulates the autophagic activity of p62. Mechanistically, p62 acetylation at K420 and K435 impairs the UBA domain homodimerization and enhances its affinity to ubiquitin, but does not affect the affinity of p62 to LC3, thus inducing the p62 body formation and its autophagic degradation and contributing to cell survival under nutrient stress [18]. Our experiments revealed that UFM1 failed to affect either p62 dimerization or its association with ubiquitinated proteins. Notably, expression of UFM1 increased the p62 UFMylation and promoted its binding to LC3. In contrast, p622KR weakened its interaction with LC3 while p622KR-UFM1ΔC4 restored this interaction. This differs from the acetylation at the same site on p62, a modification that has been previously reported [18].
Interestingly, it has been reported that S-acylation of p62 at Cys289/Cys290 potentiates its association with LC3-positive membranes and facilitates the engulfment of p62 condensates, thereby augmenting selective autophagic flux [61]. In line with this, inhibition of the deacylating enzyme acyl protein thioesterase 1 ameliorates the HD pathology by restoring brain palmitoylation levels [62]. Recent studies have demonstrated that palmitoylation of p62 is essential for autophagy and that this process is impaired in both the brains of HD patients and YAC128 mice, which is reminiscent to p62 UFMylation [14]. These observations corroborate our hypothesis that upregulating p62-mediated selective autophagy represents a viable therapeutic strategy for neurodegeneration. Conversely, not all PTMs that enhance the p62-LC3 interaction are beneficial. For instance, elevated nitric oxide levels induce p62 S-nitrosylation at Cys331, which increases the binding of p62 to LC3 but inhibits autophagic flux. This disruption to autophagic flux contributes to the aggregation and dissemination of α-synuclein in Parkinson's disease and Lewy body dementia. Presumably, this impairment stems from the excessive affinity of S-nitrosylated p62 for LC3II, which competitively disrupts the recruitment of fusion machinery to the autophagosomal outer membrane [63].
In this work, we discovered that UFMylation, a previously unrecognized PTM on p62, is critical for the function of p62 in autophagic degradation. However, it remains unclear how p62 UFMylation is dynamically modulated and whether UFMylation intertwines with other modifications under physiological, stressed, or diseased conditions. Further studies are required to answer these questions. Nevertheless, this work paves a new avenue to exploring the diverse biological functions of p62 at the PTM level.
Materials and Methods
Reagents and antibodies
Bafilomycin A1 (Baf A1, HY-100558) was purchased from MedChemExpress (USA). Biotin (V900418), chloroquine (CQ, C6628), and DAPI (D9542) were ordered from Sigma-Aldrich (USA). Antibodies were obtained from the following sources: anti-p62 antibody (P0067), anti-polyQ specific monoclonal antibody (MABN2427), and anti-UFL1 antibody (HPA030559) from Sigma-Aldrich; anti-p62 antibody (sc-28359) and anti-ubiquitin antibody (Ub, sc-8017) from Santa Cruz Biotechnology (USA); anti-UFM1 antibody (ab109305) and anti-UFSP2 antibody (ab185965) from Abcam (UK); anti-LC3B antibody (A19665) from ABclonal (China); anti-beclin-1 antibody (20543-1-AP), anti-UBA5 antibody (12093-1-AP), anti-FLAG antibody (20543-1-AP), anti-HA antibody (51064-2-AP), anti-Myc antibody (60003-2-Ig), and anti-GAPDH antibody (60004-1-Ig) from Proteintech Group (China); anti-DARPP32 (2306) antibody from Cell Signaling Technology (USA); Alexa Fluor 488 goat anti-rabbit IgG (AZ1206), Alexa Fluor 488 goat anti-mouse IgG (A11001), Alexa Fluor 594 goat anti-rabbit IgG (A11012), Alexa Fluor 594 goat anti-mouse IgG (A11005), Alexa Fluor 647 goat anti-mouse IgG (A22728), and Alexa Fluor 647 goat anti-rat IgG (A21246) from ThermoFisher Scientific (USA).
Plasmid construction
Human UBA5, UFC1, UFL1, and UFBP1 were amplified from a cDNA library and cloned into pcDNA3.1-Myc or pcDNA3.1-HA vector. Human and murine p62 were obtained from the corresponding cDNA libraries and cloned into the pHBLV-FLAG, pcDNA3.1-FLAG, and V101-FLAG vectors. UFM1 was cloned into the pHBLV-His6-BAP or pcDNA3.1-His6 vector. Mutagenesis was performed using a previously described method [64]. The shp62 sequences were cloned into the pLKO.1-TRC vector (shp62 Forward oligo: 5'-CCGGGGCCTACCTTCTGGGCAAGGACTCGAGTCCTTGCCCAGAAGGTAGGCCTTTTTG-3', Reverse oligo: 5'-AATTCAAAAAGGCCTACCTTCTGGGCAAGGACTCGAGTCCTTGCCCAGAAGGTAGGCC-3'). mCherry-GFP-LC3 [50] and GFP-Htt60Q plasmids [53] were kindly provided by Dr. Guanghui Wang from Soochow University.
siRNA and plasmid transfection
siRNA sequences used in this work were: siUBA5#1: 5'-CGUACCUUUGCCGUAGCAA-3'; siUBA5#2: 5'-CUGAAACGAGAAGGTGUUU-3'. siUFL1#1: 5'-GGAACUUGUUAAUAGCGGA-3'; siUFL1#2: 5'-GAGGAGUAAUUUUUACGGA-3'. HeLa and MCF-7 cells were transfected with siNC (160818) or target siRNA using riboFECTTM CP transfection Kit (C1051105, RiboBio, China) according to a previous publication [65]. Lipofectamine™ 2000 transfection reagent (11668019, Invitrogen, USA) was used to transfect plasmids into cells.
Cell culture
HEK293T, HeLa, and MCF-7 cells were obtained from American Type Culture Collection (ATCC, USA). HEK293T-sgUFSP2 and HEK293T-sgCtrl cells were kindly provided by Dr. Yu-sheng Cong from Hangzhou Normal University [28]. Cells were cultured in DMEM or RPMI 1640 supplemented with 10% FBS, penicillin, and streptomycin, and placed in a cell culture incubator with 5% CO2 and full humidity at 37 °C.
Cortical neuron culture
The C57BL/6 mouse brains from embryos at E16 were aseptically collected. The cerebral cortices were isolated under a microscope and digested with 0.25% trypsin, which was stopped by adding Neurobasal medium (21103049, Gibco) supplemented with DNase I (5 mg/mL, DN25, Sigma-Aldrich). Cells were separated and cultured in Neurobasal medium containing 2% B-27 (17504044, Gibco), 2 mM L-glutamine (25030164, Gibco), and 50 U/mL penicillin and streptomycin. Then, the primary cortical neurons were cultured in poly-L-lysine-coated plates (0.1 mg/mL) for one week before subsequent experiments.
Lentiviral infection
HeLa cells were treated with lentiviral particles and supplemented with 10 μg/mL polybrene for 12 h. The p62 knockdown stable cell lines were selected with puromycin for two weeks, and the knockdown efficiency of target genes was examined by immunoblotting. The primary cortical neurons were infected with lentivirus for 48 h for subsequent experiments.
Quantitative PCR (qPCR)
qPCR was performed according to a published method [66]. Primers with the following sequences were synthesized by GENEWIZ Biotech (China). LC3B Forward: 5'-GGATATAGGTCACCCCTCAG-3', Reverse: 5'-GTTAAAGGAGTTCCTGTCACC-3'; GAPDH Forward: 5'-TGCACCACCAACTGCTTAGC-3', Reverse: 5'-ACAGTCTTCTGGGTGGCAGTG-3'. GAPDH was used for normalization.
Affinity purification
Anti-FLAG affinity gel (B23102, Bimake, USA) was used to purify FLAG-tagged proteins following a previous approach [67]. Cell lysate was incubated with prewashed anti-FLAG affinity gel at 4 °C overnight. The gel was washed thrice with RIPA buffer. Proteins were eluted twice with 200 µg/mL FLAG peptide and combined for subsequent analysis.
Ni-NTA resin (30410, QIAGEN, Germany) and NeutrAvidin agarose resin (29201, ThermoFisher Scientific, USA) were used sequentially to purify the UFMylated proteins based on the previously published procedure [68]. Cell lysates were incubated with Ni-NTA resin under denaturing conditions. Purified proteins were eluted with 300 mM imidazole. Ni-NTA eluate was subsequently incubated with NeutrAvidin agarose resin at 4 °C overnight followed by washing. Proteins were eluted as described previously [64].
Protein sample preparation and MS analysis
Protein samples for MS analyses were processed as previously described [68]. Proteins were resolved by SDS-PAGE and gel bands were excised into small pieces. Proteins were trypsinized in-gel after reduction and alkylation. Peptide samples were subjected to an Orbitrap Fusion Lumos mass spectrometer equipped with an electrospray ionization inlet (ThermoFisher Scientific) as described previously [69].
LC-MS/MS raw files were analyzed with Proteome Discoverer (ThermoFisher Scientific) against the human protein database, containing decoy protein sequences and common contaminants. The methionine oxidation, N-terminal acetylation, and UFM1 remnant (Val-Gly, obtained after trypsin cleavage of the peptide bond after Arg near the UFM1 C-terminus) on Lys were set as variable modifications. Log2 (Fold Change) (UFM1/pHBLV) was obtained based on the protein abundance in the pHBLV-His6-BAP-UFM1 and pHBLV expressing samples. P-value was calculated with a two-tailed Student's t-test for data from three biological replicates. Volcano plot was constructed in Origin 9 using Log2 (Fold Change) and -Log10 (P-value).
Immunoblotting
Immunoblotting was conducted following a published method [70, 71]. In brief, proteins were separated by SDS-PAGE and transferred to a piece of PVDF membrane, which was blocked, and incubated with primary and secondary antibodies. Chemiluminescent signals were imaged and analyzed using a Tanon 5200 imaging system (China).
Mouse brain tissue preparation and dissection
Twelve weeks after injection of the adeno-associated virus, mice were perfused with PBS and 4% paraformaldehyde, and then brains were cryoprotected in 30% sucrose solution. Serial brain sections with a thickness of 20 μm were prepared using a freezing microtome (Leica, CM1950, Germany).
Immunofluorescence
Immunofluorescence was performed according to established protocols [66]. Cells were fixed, permeabilized, blocked, incubated with primary antibodies overnight at 4 °C, and stained with Alexa Fluor 488 goat anti-rabbit IgG, Alexa Fluor 594 goat anti-rabbit IgG, or Alexa Fluor 647 goat anti-mouse IgG for 2 h in the dark. Primary cortical neurons were incubated with primary antibody overnight at 4 °C, and then with Alexa Fluor 594 goat anti-rabbit IgG, Alexa Fluor 488 goat anti-mouse IgG, and Alexa Fluor 647 goat anti-rat IgG for 2 h in the dark. The embedded mouse brain was dissected into 20 μm sections using a freezing microtome, and the slices were blocked and probed with primary antibodies overnight at 4 °C, and then incubated with Alexa Fluor 594 goat anti-rabbit IgG or Alexa Fluor 594 goat anti-mouse IgG for 2 h in the dark. Nuclei were stained with DAPI for 10 min. Images were taken with the Nikon A1R HD25 confocal microscope (Japan).
In vitro UFMylation assay
Purified UBA5 (2 μM), UFC1 (2 μM), UFL1 (2 μM), UFM1ΔC2 (2 μM), and GST-p62 (2 μM) or GST-p622KR (2 μM) were mixed in the reaction buffer (5 mM ATP, 0.05% BSA, 10 mM MgCl2, 50 mM HEPES, pH 7.5) and incubated at 30 ℃ for 2 h. The sample was mixed with 5 × SDS sample loading buffer and heated at 98 ºC for 10 min.
Autophagic flux assay
Autophagy flux assay was performed using a published procedure [50]. Cells transfected with mCherry-GFP-LC3 were fixed, blocked, stained with DAPI, and washed thrice with PBS. Microphotographs of mCherry-GFP-LC3 fluorescence were captured under a confocal microscope. Autophagic flux was evaluated with ImageJ by counting the number of yellow and red puncta for 30 cells. Specifically, the Analyze Particles function in ImageJ was activated, the particle size threshold was set to 300-infinity pixels2, and the Exclude on Edges option was enabled.
Sample preparation for transmission electron microscopy
Cells were fixed with glutaraldehyde (2.5%) overnight at 4 °C as described previously [72], stained with osmium tetroxide (0.1 M) in PBS for 2 h, and rinsed thrice with PBS. Then, cells were dehydrated with a gradient concentration of ethanol (30%, 50%, 70%, 80%, 85%, 90%, 95%, 100%) for 15 min each and infiltrated with epoxy resin for 12 h at 37 °C. After epoxy resin was embedded and polymerized for 48 h at 60 ºC, 80-nm-thick ultrathin sections were prepared using a diamond knife on a Leica EM UC7 ultramicrotome, and double-stained with 2% aqueous uranyl acetate and aqueous lead citrate for 15 min. Images were taken with a transmission electron microscope (Hitachi HT7700, Japan).
Animals and stereotaxic viral injection
FVB-Tg (YAC128)/Nju mice and WT littermates were purchased from Model Animal Research Center, MARC (China). B6-hHTT130-N mice (Strain #: T054804) were purchased from GemPharmatech Co., Ltd (Nanjing, China). YAC128 is a mouse model expressing the full-length human HTT gene with 128 CAG repeats. The B6-hHTT130-N mouse model incorporates a human HTT gene fragment carrying a 130 CAG repeat expansion with the mouse Htt promoter. Mice were fed with a standard chow diet and free access to water, and maintained in a standard SPF facility. Adeno-associated viruses of rAAV-hSyn-EGFP, rAAV-hSyn-mp62-P2A-EGFP, rAAV-hSyn-mp622KR-P2A-EGFP, and rAAV-hSyn-mp622KR-mUFM1ΔC4-P2A-EGFP were purchased from BrainCase (China). Eight-week-old B6-HTT-CAG130-N mice were placed on a stereotaxic instrument after being anesthetized. Adeno-associated virus (2 μL) was injected into the striatum using a 5 μL Hamilton microsyringe with an injection rate of 0.5 μL/min (anteroposterior = -1.0 mm, mediolateral = ±2.0 mm, dorsoventral = -3.3 mm from bregma). After the completion of the injection, the needle was maintained in the same position for 5 min and postsurgical care was provided for the mice. After the experiment, mice were euthanized and tissues were harvested. The animal procedure was approved by the Ethics Committee of Soochow University (SUDA20240911A28).
Statistical analysis
Data from Western blotting and qPCR were analyzed using unpaired Student's t-test or one-way ANOVA with Tukey's post hoc test for multiple comparisons. For the autophagic flux assay, statistics between multiple groups were performed using two-way ANOVA with Tukey's post hoc test in GraphPad Prism Software (San Diego, CA).
Abbreviations
ALP: autophagy-lysosome pathway; Baf A1: bafilomycin A1; CQ: chloroquine; HTT: huntingtin; HD: Huntington's disease; LC3: microtubule-associated protein 1A/1B-light chain 3; LIR: LC3-interacting region; mHTT: mutant Huntingtin; MS: mass spectrometry; MSN: medium spiny neuron; PTM: post-translational modification; UBA: ubiquitin-association; UBA5: UFM1-activating enzyme 5; UFBP1: UFM1-binding and PCI domain-containing protein 1 or DDRGK domain-containing protein 1 (DDRGK1); UFC1: UFM1-conjugating enzyme 1; UFL1: UFM1-protein ligase 1; UFSP1/2: UFM1 specific proteases 1/2; WT: wild-type.
Supplementary Material
Supplementary figures.
Supplementary table 1.
Supplementary table 2.
Supplementary table 3.
Acknowledgements
We thank Dr. Yu-sheng Cong from Hangzhou Normal University for providing the sgCtrl and sgUFSP2 HEK293T cells and Dr. Guanghui Wang from Soochow University for providing the mCherry-GFP-LC3 and GFP-Htt60Q plasmids. The MS analyses were performed at the Mass Spectrometry core facility at Suzhou Medical College of Soochow University. This work was supported by the National Natural Science Foundation of China (22177085, 22477092, and 32171437), the Basic Research Program of Jiangsu (BK20250150), the Natural Science Foundation of Jiangsu Province (BK20255001), Jiangsu Key Laboratory of Drug Discovery and Translational Research for Brain Diseases (Open Project KJS2538), Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX23_3278), the Opening Foundation of State Key Laboratory of Neurology and Oncology Drug Development, Suzhou Science and Technology Development Project (SZM2023006), Suzhou Science and Education for Health Empowerment Project (ZDXM2025010), the Interdisciplinary Basic Frontier Innovation Program (YXY2304075) and the Young Physician Scientists Program of Suzhou Medical College of Soochow University, the Suzhou Key Laboratory of Geriatric Neurological Disorders (SZS2024001), the National Center for International Research (2017B01012), and a project funded by the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions.
Author contributions
Conceptualization: G.X., X.W., J-B.L., J.M.
Methodology: X.W., X.L., H.J., W.Z., R.D., L-F.H.
Investigation: X.W., X.L., H.J., L.C., L.Z., F.L.
Visualization: X.W., H.J.
Supervision: G.X.
Writing—original draft: X.W., G.X.
Writing—review and editing: All the authors
Funding acquisition: G.X., J-B.L., J.M., X.W.
Data and materials availability
The MS data were deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the iProx [73] partner repository with the dataset identifier PXD036379 (http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD036379).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Pohl C, Dikic I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science. 2019;366:818-22
2. Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682-95
3. Levine B, Kroemer G. Biological functions of autophagy genes: A disease perspective. Cell. 2019;176:11-42
4. Deretic V. Autophagy in inflammation, infection, and immunometabolism. Immunity. 2021;54:437-53
5. Zhu W, Wang X, Li Q, Jiang X, Xu G. Post-translational modifications of selective autophagy receptors: orchestrating cellular homeostasis, disease pathogenesis, and therapeutic opportunities. Cell Mol Biol Lett. 2026;31:62
6. Tabrizi SJ, Estevez-Fraga C, van Roon-Mom WMC, Flower MD, Scahill RI, Wild EJ. et al. Potential disease-modifying therapies for Huntington's disease: Lessons learned and future opportunities. Lancet Neurol. 2022;21:645-58
7. Li Z, Wang C, Wang Z, Zhu C, Li J, Sha T. et al. Allele-selective lowering of mutant HTT protein by HTT-LC3 linker compounds. Nature. 2019;575:203-9
8. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H. et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131-45
9. Isogai S, Morimoto D, Arita K, Unzai S, Tenno T, Hasegawa J. et al. Crystal structure of the ubiquitin-associated (UBA) domain of p62 and its interaction with ubiquitin. J Biol Chem. 2011;286:31864-74
10. Lee Y, Weihl CC. Regulation of SQSTM1/p62 via UBA domain ubiquitination and its role in disease. Autophagy. 2017;13:1615-6
11. Jakobi AJ, Huber ST, Mortensen SA, Schultz SW, Palara A, Kuhm T. et al. Structural basis of p62/SQSTM1 helical filaments and their role in cellular cargo uptake. Nat Commun. 2020;11:440
12. Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S. et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci. 2010;13:567-76
13. Ehrnhoefer DE, Martin DDO, Schmidt ME, Qiu X, Ladha S, Caron NS. et al. Preventing mutant huntingtin proteolysis and intermittent fasting promote autophagy in models of Huntington disease. Acta Neuropathol Commun. 2018;6:16
14. Abrar F, Davies MC, Alshehabi Y, Kumar A, Dang A, Nguyen YTN. et al. Reduced palmitoylation of SQSTM1/p62 in Huntington disease is associated with impaired autophagy. FASEB J. 2025;39:e70549
15. Ochaba J, Lukacsovich T, Csikos G, Zheng S, Margulis J, Salazar L. et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc Natl Acad Sci U S A. 2014;111:16889-94
16. Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell. 2011;44:279-89
17. Matsumoto G, Shimogori T, Hattori N, Nukina N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum Mol Genet. 2015;24:4429-42
18. You Z, Jiang WX, Qin LY, Gong Z, Wan W, Li J. et al. Requirement for p62 acetylation in the aggregation of ubiquitylated proteins under nutrient stress. Nat Commun. 2019;10:5792
19. Lee Y, Chou TF, Pittman SK, Keith AL, Razani B, Weihl CC. Keap1/Cullin3 modulates p62/SQSTM1 activity via UBA domain ubiquitination. Cell Rep. 2017;19:188-202
20. Gao K, Shi Q, Liu Y, Wang C. Enhanced autophagy and NFE2L2/NRF2 pathway activation in SPOP mutation-driven prostate cancer. Autophagy. 2022;18:2013-5
21. Shi Q, Jin X, Zhang P, Li Q, Lv Z, Ding Y. et al. SPOP mutations promote p62/SQSTM1-dependent autophagy and Nrf2 activation in prostate cancer. Cell Death Differ. 2022;29:1228-39
22. Aichem A, Kalveram B, Spinnenhirn V, Kluge K, Catone N, Johansen T. et al. The proteomic analysis of endogenous FAT10 substrates identifies p62/SQSTM1 as a substrate of FAT10ylation. J Cell Sci. 2012;125:4576-85
23. Sasakawa H, Sakata E, Yamaguchi Y, Komatsu M, Tatsumi K, Kominami E. et al. Solution structure and dynamics of Ufm1, a ubiquitin-fold modifier 1. Biochem Biophys Res Commun. 2006;343:21-6
24. Wang X, Lv X, Ma J, Xu G. UFMylation: An integral post-translational modification for the regulation of proteostasis and cellular functions. Pharmacol Ther. 2024;260:108680
25. Komatsu M, Noda NN, Inada T. The mechanistic basis and cellular functions of UFMylation. Nat Rev Mol Cell Biol. 2026: doi:10.1038/s41580-025-00944-y.
26. Yoo HM, Kang SH, Kim JY, Lee JE, Seong MW, Lee SW. et al. Modification of ASC1 by UFM1 is crucial for ERα transactivation and breast cancer development. Mol Cell. 2014;56:261-74
27. Kang SH, Kim GR, Seong M, Baek SH, Seol JH, Bang OS. et al. Two novel ubiquitin-fold modifier 1 (Ufm1)-specific proteases, UfSP1 and UfSP2. J Biol Chem. 2007;282:5256-62
28. Liang Q, Jin Y, Xu S, Zhou J, Mao J, Ma X. et al. Human UFSP1 translated from an upstream near-cognate initiation codon functions as an active UFM1-specific protease. J Biol Chem. 2022;298:102016
29. Millrine D, Cummings T, Matthews SP, Peter JJ, Magnussen HM, Lange SM. et al. Human UFSP1 is an active protease that regulates UFM1 maturation and UFMylation. Cell Rep. 2022;40:111168
30. Liu J, Wang Y, Song L, Zeng L, Yi W, Liu T. et al. A critical role of DDRGK1 in endoplasmic reticulum homoeostasis via regulation of IRE1α stability. Nat Commun. 2017;8:14186
31. Walczak CP, Leto DE, Zhang L, Riepe C, Muller RY, DaRosa PA. et al. Ribosomal protein RPL26 is the principal target of UFMylation. Proc Natl Acad Sci U S A. 2019;116:1299-308
32. Simsek D, Tiu GC, Flynn RA, Byeon GW, Leppek K, Xu AF. et al. The mammalian ribo-interactome reveals ribosome functional diversity and heterogeneity. Cell. 2017;169:1051-65 e18
33. Qin B, Yu J, Nowsheen S, Wang M, Tu X, Liu T. et al. UFL1 promotes histone H4 ufmylation and ATM activation. Nat Commun. 2019;10:1242
34. Wang Z, Gong Y, Peng B, Shi R, Fan D, Zhao H. et al. MRE11 UFMylation promotes ATM activation. Nucleic Acids Res. 2019;47:4124-35
35. Ishimura R, El-Gowily AH, Noshiro D, Komatsu-Hirota S, Ono Y, Shindo M. et al. The UFM1 system regulates ER-phagy through the ufmylation of CYB5R3. Nat Commun. 2022;13:7857
36. Liu J, Guan D, Dong M, Yang J, Wei H, Liang Q. et al. UFMylation maintains tumour suppressor p53 stability by antagonizing its ubiquitination. Nat Cell Biol. 2020;22:1056-63
37. Yang X, Wen Y, Qin S, Zhou Y, Zhang C, Huang L. et al. UFMylation maintains tumor suppressor pVHL stability by activating the deubiquitinase BAP1. Sci Adv. 2025;11:eadt8800
38. Zou Y, Wang Z, Jiang Q, Kong X, Ma X, Liang Z. et al. UFMylation: A supervisor of the HIF1α pathway and a potential therapeutic target for anti-PD-1 combination therapy in hypoxic tumors. Proc Natl Acad Sci U S A. 2025;122:e2500562122
39. Wu X, Gao H, Zhuang Q, Li F, Feng X, Shao N. et al. Loss of UFL1 confers enzalutamide resistance of prostate tumors by regulating METTL16-mediated m6A modification of EEF1A1 mRNA. Int J Biol Sci. 2026;22:1306-21
40. Wang Z, Xiong S, Wu Z, Wang X, Gong Y, Zhu WG. et al. VCP/p97 UFMylation stabilizes BECN1 and facilitates the initiation of autophagy. Autophagy. 2024;20:2041-54
41. Jing J, Yang F, Wang K, Cui M, Kong N, Wang S. et al. UFMylation of NLRP3 prevents its autophagic degradation and facilitates inflammasome activation. Adv Sci (Weinh). 2025;12:e2406786
42. Zhang M, Zhu X, Zhang Y, Cai Y, Chen J, Sivaprakasam S. et al. RCAD/Ufl1, a Ufm1 E3 ligase, is essential for hematopoietic stem cell function and murine hematopoiesis. Cell Death Differ. 2015;22:1922-34
43. Huber J, Obata M, Gruber J, Akutsu M, Lohr F, Rogova N. et al. An atypical LIR motif within UBA5 (ubiquitin like modifier activating enzyme 5) interacts with GABARAP proteins and mediates membrane localization of UBA5. Autophagy. 2020;16:256-70
44. Cao Y, Li R, Shen M, Li C, Zou Y, Jiang Q. et al. DDRGK1, a crucial player of ufmylation system, is indispensable for autophagic degradation by regulating lysosomal function. Cell Death Dis. 2021;12:416
45. DeJesus R, Moretti F, McAllister G, Wang Z, Bergman P, Liu S. et al. Functional CRISPR screening identifies the ufmylation pathway as a regulator of SQSTM1/p62. eLife. 2016;5:e17290
46. Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, Abdellatif M, Abdoli A, Abel S. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy. 2021;17:1-382
47. Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y. et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213-23
48. Lee DH, Park JS, Lee YS, Han J, Lee DK, Kwon SW. et al. SQSTM1/p62 activates NFE2L2/NRF2 via ULK1-mediated autophagic KEAP1 degradation and protects mouse liver from lipotoxicity. Autophagy. 2020;16:1949-73
49. Peng H, Yang F, Hu Q, Sun J, Peng C, Zhao Y. et al. The ubiquitin-specific protease USP8 directly deubiquitinates SQSTM1/p62 to suppress its autophagic activity. Autophagy. 2020;16:698-708
50. Zhang C, Huang Z, Huang X, Ma Y, Cao Y, Zhang Z. et al. PLK2 disrupts autophagic flux to promote SNCA/α-synuclein pathology. Autophagy. 2025;21:1623-43
51. Jin J, Meng X, Huo Y, Deng H. Induced TRIM21 ISGylation by IFN-β enhances p62 ubiquitination to prevent its autophagosome targeting. Cell Death Dis. 2021;12:697
52. Sterner DE, Nathan D, Reindle A, Johnson ES, Berger SL. Sumoylation of the yeast Gcn5 protein. Biochemistry. 2006;45:1035-42
53. Zhou L, Hao Z, Wang G, Xu G. Cereblon suppresses the formation of pathogenic protein aggregates in a p62-dependent manner. Hum Mol Genet. 2018;27:667-78
54. Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y. et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555-67
55. Van Raamsdonk JM, Pearson J, Slow EJ, Hossain SM, Leavitt BR, Hayden MR. Cognitive dysfunction precedes neuropathology and motor abnormalities in the YAC128 mouse model of Huntington's disease. J Neurosci. 2005;25:4169-80
56. Sun H, Li M, Li Y, Zheng N, Li J, Li X. et al. Gastrodin improves the activity of the ubiquitin-proteasome system and the autophagy-lysosome pathway to degrade mutant Huntingtin. Int J Mol Sci. 2024;25:7709
57. Pouladi MA, Morton AJ, Hayden MR. Choosing an animal model for the study of Huntington's disease. Nat Rev Neurosci. 2013;14:708-21
58. Lamark T, Johansen T. Mechanisms of selective autophagy. Annu Rev Cell Dev Biol. 2021;37:143-69
59. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349-64
60. Zhu J, Ma X, Jing Y, Zhang G, Zhang D, Mao Z. et al. P4HB UFMylation regulates mitochondrial function and oxidative stress. Free Radic Biol Med. 2022;188:277-86
61. Huang X, Yao J, Liu L, Chen J, Mei L, Huangfu J. et al. S-acylation of p62 promotes p62 droplet recruitment into autophagosomes in mammalian autophagy. Mol Cell. 2023;83:3485-501 e11
62. Virlogeux A, Scaramuzzino C, Lenoir S, Carpentier R, Louessard M, Genoux A. et al. Increasing brain palmitoylation rescues behavior and neuropathology in Huntington disease mice. Sci Adv. 2021 7
63. Oh CK, Dolatabadi N, Cieplak P, Diaz-Meco MT, Moscat J, Nolan JP. et al. S-Nitrosylation of p62 inhibits autophagic flux to promote α-Synuclein secretion and spread in Parkinson's disease and Lewy body dementia. J Neurosci. 2022;42:3011-24
64. Wang X, Cao L, Jiang H, Zhou L, Hu Z, Xu G. Proximity proteomics and biochemical analysis reveal a noncanonical function for UFM1-specific protease 1 in the p62 body formation. J Proteome Res. 2023;22:2352-63
65. Yu W, Wang B, Zhou L, Xu G. Endoplasmic reticulum stress-mediated p62 downregulation inhibits apoptosis via c-Jun upregulation. Biomol Ther (Seoul). 2021;29:195-204
66. Duan CY, Li Y, Zhi HY, Tian Y, Huang ZY, Chen SP. et al. E3 ubiquitin ligase UBR5 modulates circadian rhythm by facilitating the ubiquitination and degradation of the key clock transcription factor BMAL1. Acta Pharmacol Sin. 2024;45:1793-808
67. Xu YJ, Zeng K, Ren Y, Mao CY, Ye YH, Zhu XT. et al. Inhibition of USP10 induces myeloma cell apoptosis by promoting cyclin D3 degradation. Acta Pharmacol Sin. 2023;44:1920-31
68. Lu J, Jiang H, Li D, Chen T, Wang Y, Pu Z. et al. Proximity labeling, quantitative proteomics, and biochemical studies revealed the molecular mechanism for the inhibitory effect of indisulam on the proliferation of gastric cancer cells. J Proteome Res. 2021;20:4462-74
69. Zhu Y, Lei Q, Li D, Zhang Y, Jiang X, Hu Z. et al. Proteomic and biochemical analyses reveal a novel mechanism for promoting protein ubiquitination and degradation by UFBP1, a key component of ufmylation. J Proteome Res. 2018;17:1509-20
70. Guo DK, Zhu Y, Sun HY, Xu XY, Zhang S, Hao ZB. et al. Pharmacological activation of REV-ERBα represses LPS-induced microglial activation through the NF-κB pathway. Acta Pharmacol Sin. 2019;40:26-34
71. Zhou L, Huang X, Niesvizky R, Pu Z, Xu G. Caspase-8 regulates the antimyeloma activity of bortezomib and lenalidomide. J Pharmacol Exp Ther. 2021;379:303-9
72. Wang X, Zhu YT, Zhu Y, Sun YL, Huang J, Li Z. et al. Long-term running exercise alleviates cognitive dysfunction in APP/PSEN1 transgenic mice via enhancing brain lysosomal function. Acta Pharmacol Sin. 2022;43:850-61
73. Ma J, Chen T, Wu S, Yang C, Bai M, Shu K. et al. iProX: An integrated proteome resource. Nucleic Acids Res. 2019;47:D1211-D7
Author contact
![]() Corresponding authors: Jingjing Ma (jingjingmajjedu.cn), Jia-Bin Li (lijiabinedu.cn), and Guoqiang Xu (gux2002edu.cn).
Corresponding authors: Jingjing Ma (jingjingmajjedu.cn), Jia-Bin Li (lijiabinedu.cn), and Guoqiang Xu (gux2002edu.cn).